Department of Chemical Engineering, Instituto. Superior Técnico, Lisbon (Portugal), October 1997
http://www.ist.utl.pt/
1. Structure, properties, classification and analysis of lipids
2. Sources and composition of oils and fats
3. Processing of raw materials
4. Deterioration of oils, fats and emulsions
Structure, properties and classification of lipids.
Edible oils and fats sources, processing, bulk products and dispersions.
Chemical, biochemical and biological deterioration.
1. Structure, properties, classification and analysis of lipids.
Lipids are a group of compounds exhibiting intermediate to low polarity and low water solubility, which constitute one of the three main components of living organisms. One of their peculiarities is that when metabolized, they are capable of generating more than twice the amount of energy obtained from equivalent weight of carbohydrates (contributing ca. 39 kJ/kg). Though a large majority of lipids are fatty acid triesters of glycerol, the other lipids' importance is out of proportion with their relative abundance, because of their structural roles, namely at the interfaces separating hydrophylic and hydrophobic parts of the tissues and organs.
Fats and oils are triacylglycerols separated by the slightly arbitrary distinction of solid or liquid state at room temperature. In triacylglycerols of vegetable origin, fatty acids esterified onto position 2 significantly differ from those esterified onto positions 1 and 3 - which exhibit little overall difference in substitution pattern, whereas in products of animal origin random substitution seems to predominate.
Lipids exhibit peculiar melting properties which form the basis for their selection for a number of uses and underlie apparent physiological selectivities. These will be dealt with in section 3.5 .
Lipid Class Composition
Acylglycerols Mono-, di- and triesters of Glycerol
Waxes Fatty acid esters of fatty alcohols
Phosphoacyl glycerols Diacylglycerol and an amine or alcohol esterified to phosphoric acid
Sphyngomielins Sphingosine plus a fatty acid esterified to phosphatidylcholine
Cerebrosides Sphingosine , fatty acid and a hexose
Gangliosides Sphgingosine, a fatty acid and a sialic acid carbohydrate
Steroids C30 Tetracyclic triterpenes
Carotenoids C40 polyunsaturated tetraterpenas
Tocols Tocopherols and tocotrienols, phenolic antioxidants including vitamin E
Other Phytomenadione,...
Analysis of lipids is usually performed by gas chromatography after selective extraction normally performed with chloroform/methanol (2:1 by volume), for instance by thin layer chromatography.
Oil and fat analysis includes routinely the determination of unsaturation, free acidity, unsaponifiable materials, and fatty acid profile, and may include stereospecific analysis. Chromatographic determination of actual triacylglycerols, either by HPLC or GLC methods, is also usual when dealing with solid fats or when analysing oils in search of their genuine origin. In this respect it might also be useful to determine the stereochemical configuration of carbon 2 of glycerol - characterizing the populations of positions 1 and 3 separately in order to find out whether significant differences exist. This may be achieved using a combination of chemical and enzymatic analytical methodologies, which yields the absolute configuration around the carbon atom 2 of glycerol. For this purpose, pancreatic lipase hydrolysis is carried out under controled mild conditions yielding a mixture of 1,2-diacylglycerols, 2,3-diacylglycerols and fatty acids characteristic of positions 1 and 3. Under stronger conditions the 2-acylglycerol is obtained. Fatty acid separation and analysis yields the composition at these two positions, 1+3. The mixture of diacylglycerides from the first reaction is then subject to diacylglycerol kinase phosphorilation, which occurrs given ATP, but specifically at position 3 of the 1,2-diacylglycerol. The phosphorylated diacylglycerol is separated from the 2,3-diacylglycerol. Fatty acid profiles are obtained after chemical ester hydrolysis, derivatisation and chromatography, for position 2 (from the 2-acylglycerol), for 1+2, from the phosphorylated compound, yielding the composition for position 1 by difference, and for position 3 from the 2,3-diacylglycerol, and also from the composition obtained for the fatty acids resulting from lysis of 1+3 ester bonds when preparing the 2-acylglycerol. Selective hydrolysis using some of the phospholipases may also be used to characterize fatty acid composition at position 2 or at position 3.
Unsaturation is usually given as the iodine value, a numerical quantity proportional to the weight of iodine which adds to a given quantity of lipid (generating viccinal diiodo compounds from pre-existing double bonds). For this purpose iodine in solution is allowed to add to the lipid, and the excess is back titrated with thiosulphate.
Free acidity measures the amount of free acids present in the sample, and may further distiguish between free fatty acids and inorganic acidity.
Unsaponifiable content is measured to determine the total content of non acylglycerol material, and is a quantity usually specified for oils and fats.
Fatty acid profile determination is performed by GLC after either saponification and neutralization or, more recently, acid hydrolysis of the oil. In the latter, BF3 or acetyl chloride may be used to liberate the fatty acids.
Physical properties of fats are extremely important. They are the consequence of the triacylglycerol composition, which influences the nature, stability and structure of ordered phases. Influences are felt which relate to the number of carbon atoms, unsaturation and conformation of the fatty acids, others mainly to the triacylglycerol structure itself.
Even numbered fatty acid moieties, the most abundant ones throughout, tend to show higher solid/liquid transition temperatures due to higher Van der Waals interactions per unit weight. Saturated fatty acid residues tend to adopt mainly a zigzag type of conformation when in the crystal lattice of triacylglycerols, and the inclusion of a trans double bond does not significatively affect this situation, despite the slight shortening of the carbon chain which it entails. The existence of a cis double bond introduces a bending of the chain, and lowers transition temperatures. Much the same situation arises in the somewhat simpler situation of fatty acid crystals, themselves, and is illustrated by the relative values of stearic acid (69.6 °C), which is the saturated 18 carbon atom fatty acid often abbreviated as (18:0), which compares with 62.9 ºC for palmitic acid (16:0) and only 61.3 for margaric acid (17:0), and also with 13.4 °C for oleic acid (18:1, (9-cis)), and 46 °C for elaidic acid (18:1, (9-trans)). This feature is even stronger when various isolenic cis double bonds are present,as in ![]() -linoleic acid (18:2, (9-cis, 12-cis) which melts at -5 °C and
-linoleic acid (18:2, (9-cis, 12-cis) which melts at -5 °C and ![]() -linolenic acid (18:3, (9-cis, 12-cis, 15-cis)), which melts at -11 °C.
-linolenic acid (18:3, (9-cis, 12-cis, 15-cis)), which melts at -11 °C.
A special positional identification system for the cis double bonds is commonly used for lipids.
This system is based upon the distance to a double bond when starting from the methyl group end of the fatty acid. Thus ![]() 3 C18 acids are those which have a double bond linking carbons 15 and 16, such as
3 C18 acids are those which have a double bond linking carbons 15 and 16, such as ![]() -linolenic acid
-linolenic acid
Despite the fact that this nomenclature differs from the IUPAC nomenclature, it is useful especially because the number of carbon atoms which separate the methyl group from the double bond is related to the physiological role of the fatty acid, apparently, and also indicates common biosynthesis. The role of ![]() 6 acids such as linoleic (9-cis,12-cis-octadienoic), and
6 acids such as linoleic (9-cis,12-cis-octadienoic), and ![]() -linolenic 6-cis,9-cis,12-cis-octatrienoic) is different from that of eicosapentaenoic acid (6-cis,9-cis,12-cis,15-cis, 18-cis -eicosapentaenoic acid) or of its twenty two carbon atom homologue docosahexaenoic acid, both of which are important fish oil components and which belong to the
-linolenic 6-cis,9-cis,12-cis-octatrienoic) is different from that of eicosapentaenoic acid (6-cis,9-cis,12-cis,15-cis, 18-cis -eicosapentaenoic acid) or of its twenty two carbon atom homologue docosahexaenoic acid, both of which are important fish oil components and which belong to the ![]() 3 family together with
3 family together with ![]() -linolenic acid. Oleic acid (9-cis-octenoic acid) belongs to the group of
-linolenic acid. Oleic acid (9-cis-octenoic acid) belongs to the group of ![]() 9 acids.
9 acids.
The patterns of distribution of the various fatty acid residues among the glycerol positions also critically affect the crystallisation behaviour, polymorfism, compatibility and rheology of each particular fat. This topic bears importance, for instance in determining the plausibility of using a given triacylglycerol moiety as a replacement for the expensive cocoa butter, but outside the scope of this work.
2. Sources and composition of oils and fats.
Oils and fats used by mankind are diverse in their origins and composition. The distinction between fats and oils (that of physical state at the arbitrary room temperature value of 20 °C) is not scientifically meaningful and these terms will therefore be used interchangeably. Mention should nevertheless be made that with the exception of fish oils and cocoa butter, most animal sources yield fats and most vegetable ones yield oils.
Main sources of oil for humanity are in constant change as new vegetable cultures and cultivars evolve replacing older ones, and as diverse consumer needs and industrial purification technologies are developed. Soyabean is the main source of vegetable oil nowadays, but fragile in a market acception, inasmuch as only ca. 19% by weight of this seed is extractable as oil, whereas oil constitutes 50% of peanut or ca. 47% of sunflowerseed.
Vegetable sources of oils are both annual cultures (such as sunflower or soyabean) and perenial (such as palm and olive trees), and oil accumulates both in seeds (as palm kernel and cottonseed) and in fruits (such as avocado and coconut). Animal sources range from the menhaden fish caught off the US Southeaster cost, to lard hogg, bovine, buffalo or other domesticated animals around the world, and also to seals caught in the artic areas.
The physiological necessity of keeping lipids liquid, and the absence of internal temperature regulation mechanisms except for warm blooded animals may mean that more unsaturated fats are obtained in colder climates, even using the same cultivar. This characteristic is especially noticeable with sunflower which may be predominantely oleic when subject to high temperature cultivation, or predominantly linoleic under more usual temperature conditions.
Annual per capita consumption of oils and fats exhibits a pronounced statistical dependence from the affluence level, with variable dependence from specific cultural or regional pReferences. Though nutritional factors are complex and much controversy still surrounds the consumption of fats, it may be conservatively stated, as the World Health Organisation does, that no more than 30% of total energy be obtained from fatty components of the diet (which means no more than ca. 12% of total dry weight), of which roughly one each should be saturated, monounsaturated and polyunsaturated (and of these, roughly half should be ![]() 3 and another half
3 and another half ![]() 6).
6).
When annual yearly production of oils is examined, it is apparent that an increase of annual consumption is occurring, together with population increase, that palm oil is becoming increasingly important, having overtaken sunflowerseed in 1981, and that rapeseed production presents the maximum overall growth rate, having overtaken sunflowerseed in 1987. Rapeseed, the major cold climate oilseed benefited from strongly motivated breeding and genetic improvement work performed both in Canada (responsible for the CanolaTM cultivars) and in Northern Europe (yielding the "0" and "00" cultivars, the numerals referring to low erucic acid in oil and to additional low glucosinolate level in the oil extraction presscake).
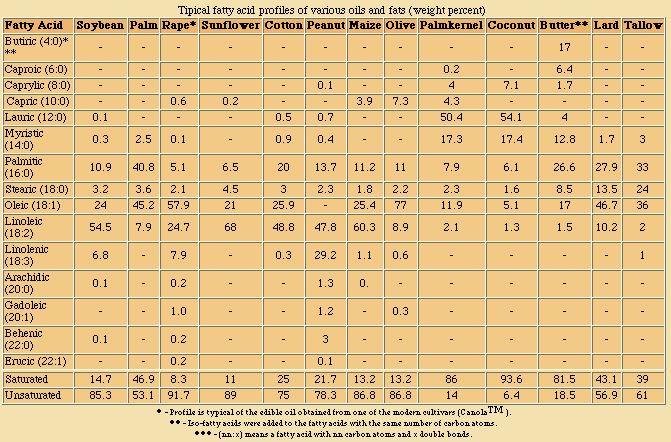
Typical fatty acid profiles of various oils and fats (weight percent)
From the profiles presented above and natural variability, it may be seen that any overall faty acid composition may be obtained for a blend of triacylglycerols. This may not be the case however for the triacylglycerol moieties themselves, and incompatibilities may become obvious upon crystallisation, when different habits may result in spontaneous fractionation.
3. Processing of raw materials:
3.1. Extraction - production of virgin oils.
Extraction of vegetable oils may be the result of simple mechanical stress and in this case it may be designated as compression, expression or expelling, or of a combination of such a treatment with a solvent-laden step. That is the general case, but solvent-free exceptions deserve special mention, as they are bound to yield oils which, not having withstood any untoward chemical or physical stimulus are named virgin oils, supposedly reflecting the composition of the naturally occurring vegetable lipid phase.
Inactivation of potentially damaging lipid breakdown catalysts, namely enzymes must precede the mechanical operation freeing the lipid from cellular enclosures. For this purpose it usual to use steam or hot water in what amounts to a blanching operation and will necessarily mean contact of a water phase with the expelled oil.
The nature of the liquid mixture is such that phase separation may become a real problem, and advantage must be taken of the enhanced separability occurring at higher temperature. One consequence of this technology is that a small percentage of hydrolysis is wont to occur, yielding free fatty acids and diglycerides as impurities.
Virgin oils are those which result from extraction operated without chemicals, and not subject to purification beyond physical phase separation at moderate temperatures.
Though virgin oil from seeds are known, commercially important virgin oils are obtained from fruit. Olive oil originating in the Mediterranean area and palm oil traditionally obtained in equatorial Africa are god examples of virgin fruit oils.
Olive oil, obtained from the expression of olives, is a highly monounsaturated oil. Composition may include as little as 0.5 % stearic acid and as high as 80% oleic, with palmitic and linoleic also relatively important. Prime material may be sold as "Extra-virgin" when less than 1% acidity (as free oleic acid), or as "Virgin" olive oil if titrating to less than 3.3% oleic acid. Higher acidity olive oils are not generally sold before refining. Virgin olive oil commands a market value which is ca. five times that of refined vegetable oils, and enjoys an upsurge of demand, as a consequence of its healthy image.
For high quality virgin oil it is of great importance that fruit does not suffer stress, both mechanical or chemical. Endogenous lipoxygenase and lipase enzymes are abundant at the fruit surface and must not be allowed contact with the oil for they will initiate oxidative and hydrolytic processes, and the reaction products will mostly remain in the oil and become responsible for its instability. Oxidation will increase PV (peroxide value) and hydrolysis will increase acidity, both contributing to a decrease in conservation of oil. It is therefore usual to pick fruit carefully avoiding damage to its skin, to expell the oil soon after harvest minimizing contact time and after decanting to keep this oil in tight containers away from oxygen and light.
Other virgin oils pale in importance when compared with olive oil. Palm oil which is deeply orange coloured due to the presence of important quantities of b-carotene is an important food item in equatorial Africa, but as an industrial commodity originates nowadays mainly from South East Asia.
3.2. Rendering - production of animal fats.
Animal fats are important materials for a number of industries, among which the production of soap and shortenings, besides their use as food items, as is the case of lard used as a frying seasoining and heat transfer medium in cooking and also as an ingredient in meat preservers, as is also the case for duck fat.
Fat occurs in animal tissues, part of it in a not very apparent way. Recovery of fat from slaughterhouse refuse is usually done by applying to it a heat treatment which melts it allowing gravity separation from non-fat materials. This treatment may be applied in heated vats or by means of continuous cooker-extruder type technology.
Fats obtained in this way may be subject to fractionation in order to obtain higher and lower melting phases, or used as such. Tallow, a by product from beef, is an important commodity, but tallows obtained from other types of cattle are used in the areas of the world where these are grown. The fat obtained from pork is called lard and it is an ingredient which is traditionally used by many, though eschewed by others.
Animal fats which are obtained after slaughter may be refined but their limited unsaturation and absence of sensitizers makes them less subject to oxidative deterioration. They may thus be used as such, eventually after addition of an antioxidant.
3.3. Butter production
Fat may be obtained from mammal’s milk, and was traditionally used by the people who kept cattle herds. It is obtained from milk fat by churning it, a mechanical process which involves heavy mixing at cool temperatures, and results in the separation of a yellowish watery emulsion containing some protein which is called buttermilk and the creation of an emulsion the continuous phase of which is lipidic. This emulsion contains aproximately twenty percent water and exhibits a plastic behaviour at room temperature, but becomes less and less spreadable as crystals of triacylglycerol grow when temperature is lowered.
Butter is an important source of energy in the diet. A yellow (carotenoid) spread it may and often contains added salt which acts not only as a flavour enhancer but also as an antimicrobial.
A relatively high valued spread, butter was the target of the first successfull man-made copy, and margarine has indeed succeeded in replacing it in may applications.
Butteroil is obtained by heating butter and allowing for gravity separation of the oil phase. Butteroil is nowadays the internationally traded commodity because of transportation costs and because of its increased stability to oxidation. Its conversion to butter may then be easily achieved, the final composition made to match local pReferences.
3.4. Refining - production, storage and packaging of refined oils
Oils and fats resulting from extraction are named crude due to the fact that they contain small quantities of compounds other than triacylglycerol esters. In the refinery these crude oils and fats are processed so as to remove undesirable substances in order to produce useful products.
The substances removed include free fatty acids, phospholipids, carbohydrates, proteins and their degradation products, water, pigments (carotenoids and chlorophyly mainly) and oxidized fat products. Thus the need for a number of commercial refining processes conceived to remove these substances is apparent.
There are two types of refining processes in common use today: physical and chemical refining. Similar steps are taken in both processes but with distinct objectives. Generally vegetable oils and meat fats are refined chemically, but certain vegetable oils, like coconut and palm, are physically refined for it is more economical. The choice of process relies normally on the convenience and the cost of the process.
It must be stressed that in both of these processes rather high temperatures are attained, a fact that might entail an appreciable degree of cis-trans isomerisation of polyunsaturate components, which is nutritionally objectionable. On the other hand a relatively large quantity of effluent originates at the refinery.
Considerations such as these actively concurr to make the refinery area prone to innovative practices which are presently the object of much R&D. Two examples of this are lower temperature processing making extensive use of adsorption processes which may come to be preferred, and the recycling and/or reuse of adsorbents.
a) Settling and Degumming
Settling means heating the fat and making it stand for the aqueous phase to separate and be withdrawn. In doing so water, proteinaceous material, phospholipids and carbohydrates are eliminated from the fat. In certain cases, particularly with oils containing substantial amounts of phospholipids (e.g. soybean oil), a primary treatment called degumming is performed by adding 2 to 3% water, agitating the mixture at 50ºC and removing the hydrated phospholipids by means of settling or centrifugation.
In the physical refining process, degummimg is performed by adding phosphoric acid to ensure phospholipid separation instead of water
b) Neutralization (chemical processing)
Caustic soda is used in this procedure to remove free fatty acids. It is vigorously mixed (a small excess, as determined in the lab) with the heated fat at relatively high temperature (say 60-80ºC) and left to stand for the aqueous phase to settle.
The aqueous solution that remains is known as foots or soapstock and, after separation, may be used for producing soap. Residual foots are eliminated from the neutral oil by washing it with hot water, along with settling or centrifugation.
Excess soapstock may constitute an environmental hazard and may de disposed of by taking advantage of the extremely low water solubility of calcium soap.
Thus an equimolar amount of calcium chloride may be directly added to the soapstock and prompt separation of the calcium soap by precipitation from a relatively pure saline (NaCl) solution will ensue. Calcium soaps are useful industrial ingredients, for instance as demoulding agents.
Even though the main purpose of the alkali treatment is the removal of free fatty acid, the process also produces a reduction in phospholipid and colouring matter content.
c) Bleaching
Heating the oil to 85ºC and treating it with adsorbents which are termed bleaching earths (Fuller’s earth, acid activated montmorillonite clays or activated carbon) permits an almost complete elimination of all colouring materials.
Attention should be paid to prevent oxidation during bleaching. Phospholipids, soaps and some oxidation materials are also adsorbed alongside the pigments. Filtration is then performed to remove the spent adsorbent.
Research into other types of adsorbent, including silica-based ones and composite materials, may one day result in oils which did not need deodorization, and also in materials which can be (relatively) cheaply reused or recycled.
d) Deodorization
Steam or nitrogen stream distillation under reduced pressure is used to remove volatile compounds with undesirable flavours, most of which originate from oxidation of the oil. It is believed that this treatment is also responsible for the thermal destruction of nonvolatile off-flavour substances, whereas the resulting volatiles are distilled away.The resulting deodorized oils will also have undergone partial cis-trans isomerisation of their polyunsaturated components, which is an unfortunate consequence of this operation, which may entail heating at temperatures as high as 240ºC for extended periods of time.
This treatment unfortunately also brings along the removal of some of the natural protectants of oils, the tocopherols, as well as the sterols. The addition of citric acid is often made to chelate traces of pro-oxidant metals, hence diminish their activity and impart to the oils, even at the diminished tocopherol content, additional stability.
Although it is established that refining generally improves the oxidative stability of oils because of the removal of prooxidants such as the chlorophyls, this is not always certain. As an example, crude cottonseed oil has a greater resistance to oxidation than its refined counterpart due to the larger amounts of gossypol and tocopherols present in crude oil. However, it is undeniable to state the remarkable added benefits that come from refining edible oils. The upgrading of palm oil quality is a good example. Moreover, adding to the improvements in colour, stability and flavour, powerful toxicants, such as gossypol in cottonseed oil and aflatoxins in peanut oil, are thoroughly eliminated during the refining process.
e) Storage and packaging
After deodorization, oil must be considered as an oxidisable consumer product. Quality will inexorably diminish as time goes by, and both passive and active protection may be called for.
Oil must therefore be handled, transported, stored and bottled under nitrogen in stainless steel vessels and through stainless steel pipes.
Packaging for catering or retail may be done in plastic containers (PVC is losing market share to polyester) which may either be made opaque or, according to public preference, at least yellow coloured in order to protect from the prooxidative action of light.
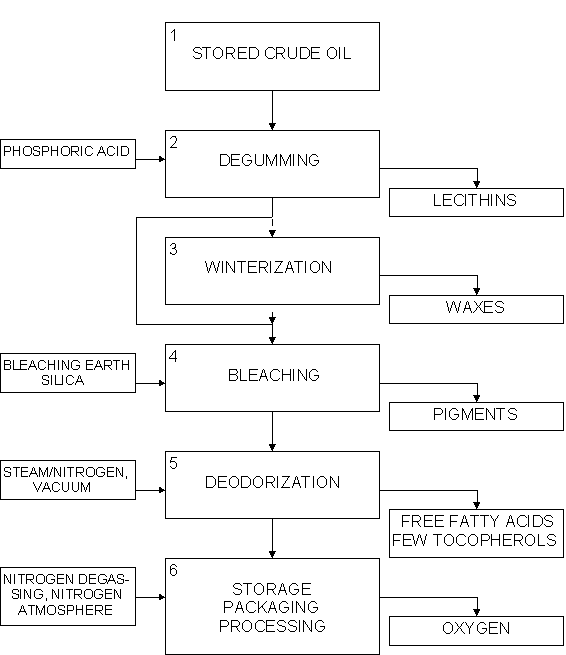
f) Process flowsheets
Physical refining
Chemical refining
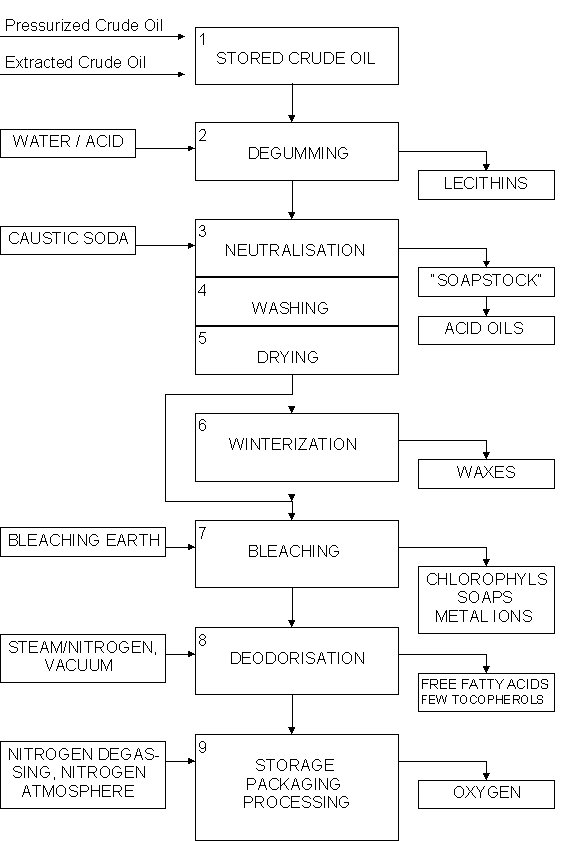
3.5. Modified oils and fats -hydrogenation, interestification and fractionation
a) Hydrogenation
Hardening of fats is produced by the addition of hydrogen to double bonds in the chains of fatty acids in triacylglycerols. This process has a vital role in the fats and oils industry because it achieves two main goals. In the first place, it permits the transformation of liquid oils into semisolid fats more indicated for specific applications, as in the cases of margarine and shortenings, and secondly, it results in materials with an improved stability.
For hydrogenation, a mixture of oil with a finely divided transition metal surface acting as a catalyst (usually nickel, but also palladium, platinium or rhodium) is heated to the hydrogenation temperature (140 to 225ºC), exposed to a hydrogen pressure of up to 60 psi whilst ensuring thorough mixing. Mixing permits faster rates of this heterogeneous reaction, helps dissipate, and is especially important given the large density difference between the catalyst and the reactants. Ensuring adequate agitation presents the most intricate technological barrier for it is mandatory that hydrogen-tightness be maintained throughout the hydrogenation device, even at moderately high pressure and when a rotor is installed, powered by an externally located engine. The starting oil must be refined, bleached, low in soap, and dry, or else the catalyst will suffer and become inactive due to preferential adsorption of any of the above (catalyst poisoning). The hydrogen must also be dry and free of sulphur, carbon dioxide and ammonia for exactly the same reason. The catalyst must possess long-term activity, act in the desired manner concerning selectivity of hydrogenation and isomer formation, and be easy to remove by filtration. Refractive index alteration, which is related to the extent of saturation of the oil, is usually used to monitor the course of the hydrogenation reaction. As the expected end point is reached, the hydrogenated oil is cooled and the catalyst filtered off.
1. Selectivity
During the hydrogenation process not only do some of the double bonds get saturated, but some may even get relocated and/or transformed from the common cis to a trans configuration. The resulting isomers are known as iso acids. Thus, partial hydrogenation may end up in the formation of a quite complex mixture of reaction products, depending on which of the double bonds are hydrogenated, what kind and degree of isomerisation occurs and the relative rates of reaction. The following diagram illustrates some reaction paths linolenate (9c,12c,15c) may go through during hydrogenation.
For natural fats the task is yet more difficult due to the complexity of the mixture of starting materials present in each. It is noteworthy that no double bond migration was considered. One important result is that when hydrogenation is complete only one saturated product is obtained, irrespective of the pathway.
Selectivity defines the relative rate of hydrogenation of the more unsaturated fatty acids when compared with that of the less saturated acids. In terms of a ratio (selectivity ratio) it is possible to obtain a quantitative measure of selectivity in more absolute terms. This ratio is expressed by the quotient of the rate of hydrogenation of linoleic acid to produce oleic acid ÷ the rate of hydrogenation of oleic acid to produce stearic acid. Constants for each reaction rate may be determined from the starting and ending fatty acid compositions and hydrogenation time. For the above reaction and under certain conditions a selectivity ratio of 12 states that linoleic acid is undergoing hydrogenation 12 times faster than oleic acid, may be typical.
The use of different catalyts as well as operating parameters and pressures will induce varied selectivities. As indicated on the following table, greater selectivity ratio (SR) values occur at high temperatures, low pressures, high catalyst concentration and a low rate of agitation. The effect of changes of processing environments upon rate of hydrogenation and on the formation of trans acids are also shown. Quite a few mechanistic speculations have been undertaken in order to explain the significance of process conditions on selectivity and rate of hydrogenation. These concepts are important whenever attempting to partially hydrogenate an oil (a process termed brush hydrogenation).The nutritional inadequacy of trans fatty acids, currently a subject of some dispute, and the fact that when led to its completion hydrogenation no longer results in any trans fatty acids has led processors to devise fat hardening systems which are based upon total hydrogenation of part of the feedstock, followed by interesterification.
In this way the fact that industrially interesting higher rates obtained at higher temperatures invariably results in high temperatures.

2. Mechanism
The mechanism of hydrogenation is thought to be the reaction between unsaturated liquid oil and atomic hydrogen adsorbed onto the metal catalyst surface. In the first place a metal complex is formed at each end of the double bond (a). This complex then reacts with an atom of catalyst-adsorbed hydrogen to form an unstable half-hydrogenated state (b or c) in which the olefin is attached to the catalyst by one link only, permitting it to rotate freely. This can now react with another hydrogen atom and separate itself from the catalyst to yield the saturated product (d) or lose a hydrogen atom to the nickel catalyst in order to restore the double bond. This regenerated double bond may find itself in the same position as in the unhydrogenated compound or in a positional and/or geometric isomer of the original double bond (e, f).
Generally it is accepted that the concentration of the hydrogen adsorbed on the catalyst is the factor that determines selectivity and isomer formation. If the catalyst is hydrogen saturated, most of the active sites hold hydrogen atoms and the probability is higher that two atoms are in position to react with any double bond upon approach. This will nevertheless result in low selectivity, because saturation of any double bond approaching the two hydrogens will ocurr promptly. However, if there are only a few hydrogen atoms adsorbed, it is likelier that only one hydrogen atom react with the double bond, producing the half-hydrogenation-dehydrogenation sequency which increases probability of isomerisation. Hereafter, operating conditions (hydrogen pressure, intensity of agitation, temperature, type and concentration of catalyst) influence selectivity by their effect on the ratio of hydrogen to catalyst sites. For example, an increase in temperature increases the speed of the reaction and produces a faster removal of hydrogen from the catalyst, thus increasing selectivity.
The possibility of being able to change the SR by altering the processing conditions permits processors considerable control over the properties of the final oil. To exemplify, a more selective hydrogenation decreases linoleic acid and improves stability, reducing the formation of fully saturated compounds and preventing excessive hardness. However, a more selective reaction will enhance the formation of trans isomers, which may present concern to those nutritionally concerned. In the past years, the search for a hydrogenation process that minimises isomerisation at the same time as it avoids the formation of excessive amounts of fully saturated material has been a main objective for manufacturers, and circumventing this problem may only be acomplished with resource to total hydrogenation plus interesterification with non-hydrogenated oil.
3. Catalysts
Catalysts vary according to the degree of selectivity that they provide. Supported on various materials, nickel is invariably used commercially to hydrogenate fats. There are however other catalysts available such as copper, copper/chromium combinations, and platinum. Palladium has been shown to be more efficient than nickel due to the amount of catalyst required, although it invariably results in greater quantities os trans isomers. Homogeneous catalysts, soluble in oil, enable greater contact between oil and catalyst, thus providing more control of selectivity, but may prove elusively difficult to separate and recover.
A number of different compounds have the capacity to poison the catalyst used, and they are often the source of problems during commercial hydrogenation processes. These include: phospholipids, sulfur compounds, partial glycerol esters, mineral acids, CO2, water and soaps.
b) Interesterification
It is of common knowledge that natural fats do not have a perfect distribution of fatty acids among the glyceride molecules. The tendency for certain acids to be more concentrated at particular sn positions varies according to species and their environment and location in the plant or animal. The physical characteristics of a fat are greatly affected not only by the nature of constituent fatty acids (i.e. chain length and unsaturation) but also by their distribution in the triacylglycerol molecules. In effect, the unique fatty acid distribution patterns of some natural fats limits their industrial applications. Interesterification is one of the processes that can be applied to improve the consistency of such fats and to improve their usefulness. This process involves rearranging the fatty acids in such a way that their distribution among the triacylglycerol molecules of the fat becomes random (random interesterification) or conforms to some special pattern (directed interesterification).
1. How does interesterification occur?
The term interesterification refers to the exchange of acyl radicals between an ester and an acid (acidolysis), an ester and an alcohol (alcoholysis) or an ester and an ester (transesterification). It is the latter reaction that is relevant to the industrial interesterification of oils (also refered to as randomization) since it involves ester interchange within a single triacylglycerol molecule (intraesterification) as well as ester exchange among different molecules.
If one considers that two (n) fatty acids (A and B) are available to be esterified to glycerol, eight possible triacylglycerol species can result (n3).
Regardless of the distribution of the two acids in the original fat (e.g. AAA and BBB or ABB, ABA, BBA), interesterification results in the "shuffling" of fatty acids within a single molecule and among triacylglycerol molecules until an equlilibrium is achieved in which all possible combinations are formed. Quantitative proportions of the different species depend only on the amount of each acid in the original fat and can be predicted.
2. Industrial Process
Interesterification results when fat is heated at relatively high temperatures (less than 200ºC) for a considerably long period. Nevertheless, catalysts are frequently used that permit a shorter period (30 min.) for the completion of the reaction at temperatures as low as 50ºC. Alkali metals and alkali metal alkylates are effective low-temperature catalysts, sodium methoxide ("methylate") being the most commonly used one. Approximately 0.1% catalyst is required. Higher concentrations may cause excessive losses of oil resulting from the formation of soap and methyl esters.
The oil to be esterified must be extremely dry and low in free fatty acids, peroxides and any other material that may react with sodium methoxide. A few minutes after the catalyst is added, the oil acquires a reddish brown color due to the formation of a complex between the sodium and the glycerides. This complex is thought to be the true catalyst. After esterification, the catalyst is inactivated through the addition of water or acid, and removed.
3. Mechanisms
There are two proposed mechanisms for interesterification, both important.
Enolate Ion Formation. This mechanism supports that an enolate ion (II), typical of the action of a base on an ester, is formed. The enolate ion reacts with another ester group in the triacylglycerol molecule to produce a ![]() -keto ester (III) which in turn reacts further with other esters to give other
-keto ester (III) which in turn reacts further with other esters to give other ![]() -keto esters. In this way all ester groups may react and in the triacylglycerol will thus move around from their initial positions.
-keto esters. In this way all ester groups may react and in the triacylglycerol will thus move around from their initial positions.
The same mode of action applies to ester interchange between two or more triacylglycerol molecules. The intra-ester ester interchange is believed to predominate in the initial stages of the reaction.
Carbonyl Addition. Here, the alkylate ion adds on to a polarized ester carboxyl producing a diglycerinate intermediate.
This intermediate reacts with another glyreride by abstracting a fatty acid, thus forming a new triacylglycerol and regenerating a diglycerinate for further reaction. Ester interchange between fully saturated S3 and unsaturated U3 molecules is shown below, as a model for the randomisation which occurs:
The K values presented are the statistical values of equilibrium constants.
4. Directed Interesterification
A random distribution is not always the most desirable. Interesterification can be directed away from randomness if the oil is maintained at a temperature below the melting point of some triacylglycerols which might result. This results in selective crystallization of the trisaturated glycerides, with the effect of removing them from the reaction mixture as they crystallize upon formation and the fatty acid composition of the liquid phase keeps changing. Interesterification thus proceeds with the formation of more trisaturated glycerides than would have otherwise occured. The process continues until most of the saturated fatty acids in the oil have precipitated. If the original oil is a liquid one containing a substancial amount of saturated acids, it is possible, by this method, to convert the oil into a mixture of products, one of them a very unsaturate oil, the other a solid fat with the consistency of shortening, obtained without resorting to hydrogenation or blending with a hard fat. The procedure is relatively slow due to the low temperature used, the time required for crystallization, and the tendency of the catalyst to become coated. A dispersion of liquid sodium-potassium alloy is commonly used to slough off the coating as it forms.
Rearrangement can also be selectively controlled during interesterification by adding excess fatty acids and continuously distilling out the liberated acids that are highly volatile. This impoverishes the fat of its acids of lower molecular weight. The content of certain acids in fat also can be reduced by using suitable solvents to extract appropriate acids during the interesterification process.
5. Applications
Interesterification finds its greatest application in the manufacture of shortenings. Lard, due to its high proportion of disaturated triacylglycerols with palmitic acid in the 2 position, forms relatively large and coarse crystals, even when scraped surface heat exchange equipment is used in order to minimize this b tendency. Shortenings made from natural lard possess a grainy consistency and poor performance in baking. Simple randomisation of lard improves its plastic range and makes it a better shortening. Directed interesterification, however, may result in a product with a higher total solids content at high temperatures and thus an extended plastic range.
Salad oil can be made from palm oil by fractionation after directed interesterification. The use of interesterification has also been applied to the production of high-stability margarine blends and hard butters that have highly desirable melting qualities, such as cocoa butter substitutes.
3.6. Emulsions and Emulsifiers
An emulsion may be perceived as a system containing two immiscible liquid phases, one of which is dispersed in the other as droplets varying between 0.1 and 50 ![]() m in diameter. The phase present in the form of droplets is said to be the internal or dispersed phase, and the matrix in which the droplets are dispersed is known as the external or continuous phase. The importance of mesomorphic or liquid crystalline phases to the properties of emulsions has been discovered only recently and is reflected in the 1972 definition of an emulsion by the IUPAC: "In an emulsion, liquid droplets and/or liquid crystals are dispersed in a liquid". The abbreviations O/W and W/O are frequently used to indicate the type of emulsion, that is, oil-in-water and water-in-oil, respectively.
m in diameter. The phase present in the form of droplets is said to be the internal or dispersed phase, and the matrix in which the droplets are dispersed is known as the external or continuous phase. The importance of mesomorphic or liquid crystalline phases to the properties of emulsions has been discovered only recently and is reflected in the 1972 definition of an emulsion by the IUPAC: "In an emulsion, liquid droplets and/or liquid crystals are dispersed in a liquid". The abbreviations O/W and W/O are frequently used to indicate the type of emulsion, that is, oil-in-water and water-in-oil, respectively.
The formation of small dispersed droplets is associated with a growth in the interfacial area between both liquids. This enlargement occurs exponentially with a decrease in droplet diameter. In specific situations the interfacial area may become unbelievably large. For example, if 1 ml of oil is dispersed as 1 mm diameter particles in water, 1.9 x 1012 globules are created and the total interfacial area is 6 m2.
The volume percentage of the dispersed phase can vary from 2-3%, in milk, to a larger value such as 65-80%, in a mayonnaise, or even to the extent of 99% in certain experimental emulsions. It is although interesting to note that perfect spheres, when packed to a maximum density, only occupy 75% of the sample volume. Consequently emulsion of dispersed-phase volumes larger than 75% only exist due to the diversity in globule size and/or the ability of the globules to deform.
The work, W, necessary to increase the interfacial area by an amount A may be represented by the relationship W = ![]()
![]() A, where
A, where ![]() is the interfacial tension. As a consequence of the large positive free energy at the interface of the two liquids, emulsions are thermodynamically unstable. Many emulsions tend to destabilise by one or more of the following three mechanisms:
is the interfacial tension. As a consequence of the large positive free energy at the interface of the two liquids, emulsions are thermodynamically unstable. Many emulsions tend to destabilise by one or more of the following three mechanisms:
Creaming or sedimentation can result from the action of gravitational force on phases that differ in density. The rate of this occurrence obeys Stokes law.
where V is the velocity of the globule, r is its radius, g is the acceleration of gravity, ![]()
![]() is the difference in density between the two phases and
is the difference in density between the two phases and ![]() is the viscosity of the continuous phase. When clustering occurs, the radius of the cluster must be used and not that of the individual components.
is the viscosity of the continuous phase. When clustering occurs, the radius of the cluster must be used and not that of the individual components.
Flocculation or clustering, which is the result of closer aggregation of globules, albeit without breach of the individual surface film, may also be the main culprit for emulsion destabilisation, as happens in non-homogenised milk.
Coalescence involves rupture of the individual globular membrane and can be the consequence of flocculation. The decrease of interfacial area makes it a very probable phenomenon as stability increases.
To produce stable emulsions, the tendency to minimise interfacial area through coalescence must be counteracted, and this is normally executed by adding emulsifiers. These are usually surface-active compounds that adsorb at the interface to lower interfacial tension to produce a physical resistance to coalescence and, occasionally, to increase surface charge.
Emulsions and emulsifiers are fundamental to the food industry. O/W emulsions come in the form of milk, cream, mayonnaise, salad dressings, ice cream mix and cake batters. On the other hand butter and margarine are W/O emulsions. "Meat emulsion" is a more complex system in which the dispersed phase is solid fat in the form of fine particles and the continuous phase is an aqueous matrix containing salts, soluble and insoluble proteins and particles of muscle fibres and connective tissues.
The ever increasing introduction of new food products and the continuing mechanisation of food processes have promoted the use of food emulsions and the need for further understanding of their properties and benefits. Emulsifying agents are nowadays commercially available, being produced to satisfy a variety of specific applications.
A. Emulsion Stability
Here are some factors that contribute towards emulsion stability:
Interfacial Tension - As indicated above, most emulsifying agents are amphiphilic compounds. They will concentrate at the oil-water interface, producing a significant reduction of the interfacial tension and will need less energy to form emulsions. Despite a lowering of interfacial tension when surface-active agents are added, the free energy of the interface remains positive, leaving a persisting state of thermodynamic instability.
Repulsion by Electric Charge - Emulsion stability is often explained by the presence of repulsive electrical charges on the surfaces of emulsion droplets. According to the DLVO theory, the dispersed particles are subject to two independent forces: the van der Waals force of attraction and the electrostatic force of repulsion arising from the presence of electrical double-layers at the particle surfaces. The net interaction between the particles is obtained by summing these two terms. If the repulsion potential exceeds the attraction potential, an energy barrier opposing collision results. If the magnitude of this energy barrier exceeds the kinetic energy of the particles, the suspension is stable. The van der Waals negative potential becomes significant only when the distance between the particles is quite small.
At intermediate distances, the repulsive potential is larger than the attractive potential. Attention should be taken on application of the DLVO theory, which was originally developed for inorganic sols (in which the dispersed phase consists of submicroscopic spherical solid particles), to emulsions (where the dispersed phase consists of oil droplets stabilised by adsorbed emulsifying agents). For example, in emulsions, coalescence involves disruption of an adsorbed film around the droplets, and calculations of the potential energy barrier opposing the collision of oil globules must take into account such factors as the distortion or flattening of the oil droplets upon close approach. However, the DLVO theory still provides a good approximation of the electrostatic contribution to emulsion stability.
Ionic surfactants contribute significantly to the stability of O/W emulsions by contributing to the establishment of electric double layers in the aqueous phase adjacent to each oil droplet. Reversibly, this mechanism is of little importance in the stabilisation of W/O emulsions, since the oil phase does not generally supply counterions in sufficient amounts to establish a strong potential gradient.
Stabilisation by Finely Divided Solids - Solid particles of very small size, as compared with the size of the dispersed oil droplet, can stabilise an emulsion by adsorbing at the interface to form a physical barrier around the droplets. In addition, energy is required to dislodge solid particles from the interface, since the oil/water interface must be increased to do so. Powdered silica, various clays, basic salts and plant cell fragments are examples of such agents.
The emulsion type produced and its stability depend largely on the relative abilities of the two phases to wet the solid particles. The phase that preferentially wets the solid particle tends to become the continuous phase. If the interfacial tension between solid and oil (![]() SO) is greater than that between solid and water (
SO) is greater than that between solid and water (![]() SW), the contact angle (
SW), the contact angle (![]() ) of the solid with the aqueous phase is less than 90º, and the major portion of the solid particle resides in the water phase, thus favouring an O/W emulsion. The converse takes place if
) of the solid with the aqueous phase is less than 90º, and the major portion of the solid particle resides in the water phase, thus favouring an O/W emulsion. The converse takes place if ![]() SW >
SW > ![]() SO. Nevertheless, if solid particles remain exclusively in either phase, they have no stabilising effect. On the other hand, the most stable emulsion is formed when the angle of contact between the two liquids and the solid surface is close to 90º. The surface of the solid and its contact angle may be modified by adjusting pH and by adsorbing various amphiphilic compounds to its surface. Concentration and chain length of the amphiphile’s hydrophobic group are important in this regard.
SO. Nevertheless, if solid particles remain exclusively in either phase, they have no stabilising effect. On the other hand, the most stable emulsion is formed when the angle of contact between the two liquids and the solid surface is close to 90º. The surface of the solid and its contact angle may be modified by adjusting pH and by adsorbing various amphiphilic compounds to its surface. Concentration and chain length of the amphiphile’s hydrophobic group are important in this regard.
Based on these considerations, it has been recommended that, for the preparation of emulsions stabilised by solid particles, a surface-active substance should be added that is soluble in the least- wetting (discontinuous) phase, and that the concentration of the surface-active agent should be adjusted to give a contact angle in the vicinity of 90º between the powder and the two liquids.
Several methods have been introduced to aid in the selection of an appropriate emulsifier, or blend of emulsifiers, for a given purpose. The most prominent of these is the one based on the relative importance of the hydrophobic and hydrophilic properties of the molecules (HLB system).
B. The HLB System for Selecting Emulsifiers
Since the principal emulsifying agents are compounds containing both hydrophobic and hydrophilic groups, and since the phase in which the emulsifier is more soluble is generally the continuous phase, the type of emulsion produced (i.e. O/W or W/O) can be predicted on the basis of the relative hydrophilic-lipophilic properties of the emulsifier. According to the hydrophilic-lipophilic balance (HLB) concept, each of the surface-active agents can be assigned a numerical value representing its hydrophilic-lipophilic balance.
Experimental determination of the HLB number for a given emulsifier is a tedious process. However, this value may be calculated with satisfactory accuracy based on easily determined characteristics of the emulsifier. The following equation was suggested by Griffin for polyhydric alcohol, fatty acid esters:
HLB = 20(1 - S/A), where S is the saponification number of the ester and A is the acid number of the acid. In certain cases, where accurate determination of the saponification number is difficult, the relationship HLB = (E+P)/5 is used, where E is the weight percent of oxyethylene and P is the weight percent of polyhydric alcohol. When ethylene oxide is the only hydrophilic group present the equation is reduced to HLB = E/5. HLB numbers for some common emulsifiers are listed below.
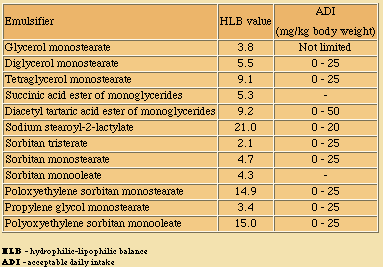
The solubility of emulsifiers in water generally follows their HLB rank. As a rule, emulsifiers with HLB values in the range 3-6 promote W/O emulsions; values between 8 and 18 promote O/W emulsions.
It has also been suggested that HLB values are algebraically additive so that the HLB of a blend of two or more emulsifiers can be obtained by simple calculation and that the blend of emulsifiers needed to produce maximum emulsion stability can be easily obtained. This, however, is not always the case. Although the HLB concept is useful as a guide of comparing emulsing-forming or stabilizing properties, it suffers from a number of limitations. First, commercial emulsifiers usually consist of a group of compounds rather than a single component. This makes direct calculation based on chemical properties very difficult. Furthermore, the HLB method does not take into consideration such factors as emulsifier concentration, mesomorphic behaviour, temperature, ionisation of the emulsifier, interaction with other compounds present, or properties and relative concentrations of the oil and aqueous phases. Pure monoglycerides, for example, have an HLB value of approximately 3.8. Accordingly they would be expected to form only W/O emulsions. However, at emulsifier concentrations that permit the formation of protective mesomorphic layers around the fat globules, pure monoacylglycerols promote O/W emulsions. Moreover, it is well known that O/W emulsions prepared from a blend of emulsifiers are usually more satble than those prepared from a single agent having the same HLB.
C. PIT as a Basis for Selecting Emulsifiers
It is obvious that temperature is an important factor in relation to the emulsion-forming characteristics of a surface-active agent. An emulsifier that tends to be preferentially soluble in water at relatively low temperatures may become preferentially soluble in oil at higher temperatures at which hydrophobic interactions become stronger. Determination of the temperature at which this inversion occurs provides a useful basis for emulsifier selection. A strong positive correlation has been observed between the phase-inversion temperature (PIT) of emulsifiers and emulsion stability.
Toxicology - Based on extensive toxicological studies, including metabolic tests and long and short term feeding experiments with animals, Acceptable Daily Intake (ADI) values have been assigned to most food emulsifiers by the FAO-WHO Codex Alimentarius Committee, and some of these values are given in the last column of the table.
Specific Food Emulsifiers - A brief discription of the most commonly used emulsifiers follows:
1. Fatty acid monoesters of ethylene or propylene glycol are also widely used in baking. A more hydrophilic ester can be prepared from a fatty acid and an alcohol, such as nonaethylene glycol.
2. Sorbitan fatty acids esters are usually mixed esters of fatty acids with sorbitol anhydride or sorbitan. Sorbitol is dehydrated first to form hexitans and hexides, which are then esterified with fatty acids. The resulting products are known commercially as "Spans".
These agents tend to promote W/O emulsions. Compounds that are more hydrophilic can be produced by reacting sorbitan esters with ethylene oxide. Polyoxyethylene chains add to the hydroxyl groups through ether linkages. The resulting polyoxyethylene sorbitan fatty acid esters are commercially known as "Tweens". In general, these compounds form hexagonal I liquid crystals in water and they can solubilise small quantities of triacylglycerols. With larger amounts of triacylglycerols, transformation to a lamellar-type liquid crystal takes place. The ability of an emulsifier to solubilise nonpolar lipids is important to the formation of phase equilibria at the emulsion interface.
3. Sodium stearoyl-2-lactylate (SSL), an ionic emulsifier, is a strongly hydrophilic surface-active agent capable of forming stable liquid crystalline phases between oil droplets and water, and thus can be used to promote very stable O/W emulsions. It is obtained from the interaction of stearic acid, 2 molecules of lactic acid, and NaOH.
Due to their strong starch-complexing abilities, sodium (and calcium) stearoyl lactates are commonly used in the baking and starch industries.
4. Phospholipids such as soybean lecithin and those in egg yolk are natural emulsifiers that promote mainly O/W emulsions. Egg yolk contains 10% phospholipid and is used to help form and stabilise emulsions in mayonnaise, salad dressing and cake. Commercial soybean lecithin contains approximately equal amounts of phosphatidylcholine, phosphatidylethanolamine and inositol. It is used to help form and stabilise emulsions in ice cream, cakes, candies and margarine. Lecithin emulsifiers of different phospholipid composition and HLB characteristics can be obtained from commercial lecithin by fractionation based on solubility in alcohol.
5. Water-soluble gums, derived from a variety of plants, are effective in stabilising O/W emulsions. They inhibit coalescence by increasing the viscosity of the continuous phase and/or by forming strong films around the oil droplets. Materials in this class include gum arabic, tragacanth, agar, pectin, xanthan, methyl- and carboxymethylcellulose and carrageenan.
6. Gylcerol esters are a class of nonionic emulsifiers extensively used in the food industry. Monoglycerides (monoacylglycerols) are prepared by direct reaction of glycerol with fatty acids or refined fats in the presence of an alkaline catalyst. Commercial monoglycerides usually contain a mixture mono-, di- and triesters of fatty acids with a monoglyceride content of about 45%. However, concentrated products containing more than 90% monoester can be prepared by mollecular distillation. Distilled monoacylglycerols are commonly used in the manufacture of margarine, snack foods, low-caloric spreads, whipped frozen dessert and pasta products.
The hydrophilic nature of a monoester can be increased by increasing the number of free hydroxyl groups in the alcoholic moeity of the molecule. Polyglycerol esters with a wide range of HLB values are thus produced by esterification of fatty acids with polyglycerols. Polyglycerol chains containing up to 30 glycerol units can be prepared by polymerisation of glycerol.
The phase behaviour of monoacylglycerol-water systems is critical for optimum functionality of monoacylglycerols in aqueous systems. With pure monoacylglycerols, the lamellar-type liquid crystal dominates for esters of the 12:0 and 16:0 fatty acids, hexagonal II or cubic type liquid crystals are usually produced from fatty acid esters with longer chains. When the water content is low, unsaturated monoacylglycerols yield lamellar-type liquid crystals at room temperature. By increasing the water content to approximately 20%, a viscous isotropic phase forms that transforms into a hexagonal II phase at temperatures above 70ºC. If the water content is increased above 40% the viscous isotropic phase will separate as gelatinous lumps, making uniform distribution very difficult.
Commercially produced distilled monoacylglycerols are frequently used in the form of aqueous mixtures to facilitate their distribution in food products. As pointed out earlier, the swelling capacity of distilled monoacylglycerols can be increased very significantly by neutralisation of the free fatty acids commonly present, or by the addition of trace amounts of ionic substances. Dilute dispersions of the commercial products, when buffered to pH 7, gave clear homogenous dispersions that form a stable gel upon cooling.
Industrial products known as crystalline hydrates are prepared by heating a mixture of about 25% saturated distilled monoacylglycerols in water to about 65ºC, acidifying the resulting mesophase with acetic or propionic acid to pH 3 and cooling with a scraped-surface heat exchanger. The product is a stable dispersion of tiny monoacylglycerol b crystals in water. The so-called hydrates, which possess unusually smooth texture, are commonly used in the baking industry.
7. The hydrophobic character of monoacylglycerols can be enhanced by the addition of various organic acid radicals yielding esters of monoacylglycerols with hydroxycarboxylic acids. Lactylated monoacylglycerols, for example, are prepared from glycerol, fatty acids and lactic acid.
Succinic and malic esters can be obtained in a similar fashion. Acetylated tartaric acid monoacylglycerols are produced by reacting the monoacylglycerol with diacetyl tartaric acid anhydride.
The diacetyl tartaric acid esters, as well as the succinic acid esters, form lamellar liquid crystals that have limited swelling capacity in water. However, as is true of distilled monoacylglycerols, their capacity to imbibe water can be increased drastically by the addition of NaOH. Malic acid esters form cubic mesomorphic phases with water contents of up to 20% and hexagonal II phases at higher temperatures and water concentrations. Succinic acid esters do not form mesomorphic phases with water, but they do exhibit mesomorphism.
3.7. Shortenings and dispersions
1. Margarine
Margarine is a fatty food manufactured in order to resemble butter in appearance, flavour, colour, texture and composition except that the fat is not principally milk fat. Margarine was invented by Hippolyte Mege-Mouriès in 1869 in France in response to need for a satisfactory butter substitute, especially for military personnel. Mege-Mouriès mixed beef fat and artificial gastric juice with salt and milk. The resultant product had pearly luster when chilled. Mege - Mouriès named this product margarine after the Greek "margarite", or pearl-like. He described the product as a variety of true butter, later as an artificial butter, butterine or margarine. Manufacture and sale of margarine was permitted by April 12, 1872, by action of the Council of Hygiene of Paris, who authorized its sale if not described as butter. Margarine manufacture expanded throughout Europe in the period 1872-4. The first American patent for margarine was granted in December 1873.
The manufacturing process evolved rapidly. Early products were formulated with artificial gastric juice and cow udder extracts. These were abandoned in favour of pasteurization of milk, dry chilling of the fat mixture and texturizing. Pasteurization of milk enabled culturing techniques to be used that improved the product flavour by more closely approaching butter flavour. Early manufacture of margarine by wet chilling of the fatty mass was replaced by dry chilling using a drum chill roll.
The history of margarine technology and formulation is a series of incremental improvements in composition and process technique. Following its invention, the major dates in margarine development are:
1890 Use of cultured milk as flavour source
1917 Cocunut oil replacement of animal fat
1923 Vitamin A addition
1934 Hydrogenated vegetable oil replacement of cocunut oil
1947 Beta carotene replacement of coal-tar colourings
1952 First soft margarine in tub form
1956 First blended vegetable/butter fat product
1957 Per capita consumption of margarine surpassd that of butter
First production of whipped stick margarine
1964 First national distribution of tub margarine
1966 Diet (40%fat) margarine"minarine" introduced
1967 Pourable fluid margarine introduced
1975 Lower fat spreads introduced
1981 Margarine/butter blends introduced nationally
2. Oil Blend Development
Margarine is a water-in-oil emulsion. An emulsion is a suspension of one liquid within a second, immiscible liquid. In margarine, the fat is the continuous phase. The dispersed phase consists of water and/or water dispersible components. The dispersible aqueous phase typically contains droplets of 1 - 20m m in diameter.
Margarine contains at least 80% fat by weight. The fat in margarine is primarily refined, mixed triglycerides (triacylglycerols) of vegetable or animal carcass origin. Marine oils are also used to formulate margarines in many countries. Tallow and lard is used in some lower-cost margarines. Lauric oils, such as cocunut and palm kernel, were used to make margarines as late as the 1930s. The predominent fat used in the United States for margarine throughout the 1970s and the 1980s has been soybean oil, followed by corn oil. In Europe and elsewhere, palm oil and its fractions are extensively used, as well as some polyunsaturate vegetable and hydrogenated marine oils.
The triacylglycerol mixture which is the main constituent of oils provides structure, lubricity and caloric density to the product. Additional fatty components may be added for their functional performance such as, for instance, lecithin or similar phosphatide which can be used to promote emulsification and retard "spatter" in frying and monoglycerides for textural smoothing and enhanced emulsification, ![]() -carotene as a colour enhancer. The oils are typically refined, bleached, partially hydrogenated and deodorised. Selectively hydrogenated oils are blended by the manufacturer to achieve required composition and physical properties for margarine.
-carotene as a colour enhancer. The oils are typically refined, bleached, partially hydrogenated and deodorised. Selectively hydrogenated oils are blended by the manufacturer to achieve required composition and physical properties for margarine.
The physical properties of margarine, especially texture, spreadability, colour, appearance and melting properties are derived from the composition of the fat and the processing technique. Margarines and similar spreads are composed of liquid oil, fat crystals and the aqueous phase. The crystals give margarine the required consistency and stabilize the water droplets. Margarines are prepared from edible oils that are chemically modified by hydrogenation and/or interesterification.
Soyabean oil and palm oil are the main alternative constituents of margarine, the US using soyabean and Europe mainly palm. Other oils as such or partially hydrogenated may be blended. These blends are useful to achieve specific ratios of solid fat to liquid fat within a range of temperatures. The standard method of measuring this ratio is the solid fat index (detailed by the American Oil Chemists Society as method Cd 10 - 57). Typical solid fat indices of margarine are shown below.
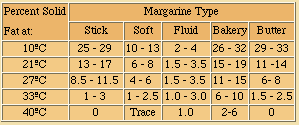
The fat crystals formed by the solid portion of the margarine fat form a three-dimensional network or lattice. Within this lattice are the liquid oil and aqueous phase of the product. As the ratio of solid-to-liquid fat increases, the texture and consistency become firmer and more rigid. Stick margarines contain 10 -15% more solid fat at 10ºC than soft or tub margarines, which cause the stick product to be more resistant to distortion.
Fats exhibit polymorphic behaviour in that they can solidify in more than one crystal form. The forms, designated ![]() ,
, ![]() ‘,
‘, ![]() , result from interaction of the component fatty acids of the tryglyceride mixture and from the rate of phase change from liquid form to solid. The
, result from interaction of the component fatty acids of the tryglyceride mixture and from the rate of phase change from liquid form to solid. The ![]() form is the least stable and lowest-melting crystal. The crystals are initially formed by the super cooling, rapid chilling process of margarine manufacture. The
form is the least stable and lowest-melting crystal. The crystals are initially formed by the super cooling, rapid chilling process of margarine manufacture. The ![]() crystals transform with time to an intermediate
crystals transform with time to an intermediate ![]() ‘ crystal that is optimal for margarine. Beta crystals can be formed under certain conditions of storage, typically from inadequate continuous refrigeration. These latter crystals show a tendency to grow and can foreshadow the development of a sandy, coarse textured product. Triacylglycerol polymorphism is not completely understood, but it is generally percieved that
‘ crystal that is optimal for margarine. Beta crystals can be formed under certain conditions of storage, typically from inadequate continuous refrigeration. These latter crystals show a tendency to grow and can foreshadow the development of a sandy, coarse textured product. Triacylglycerol polymorphism is not completely understood, but it is generally percieved that ![]() -forming tendencies are proportional to the uniformity of triacylglycerol structure.Margarine formulated for table use rather than for cooking is designed to melt completely slightly below body temperature, which prevents the mouthfeel sensation of waxiness. Puffed pastry needs margarine with the highest transition range so that melting only occurs completely after a reasonable degree of starch gelatinization. In this way the mechanically produced multilayered material initially puffs when heated and only then does lubrication by melted fat occur.
-forming tendencies are proportional to the uniformity of triacylglycerol structure.Margarine formulated for table use rather than for cooking is designed to melt completely slightly below body temperature, which prevents the mouthfeel sensation of waxiness. Puffed pastry needs margarine with the highest transition range so that melting only occurs completely after a reasonable degree of starch gelatinization. In this way the mechanically produced multilayered material initially puffs when heated and only then does lubrication by melted fat occur.
3. Emulsification and Processing
The margarine emulsion contains a dispersion of finely dispersed aqueous droplets within a crystalised fat lattice. The margarine manufacturing process consists of five unit procedures: emulsification, cooling, working, resting and packaging.
An emulsion is a suspension of one liquid in a second immiscible liquid. The margarine emulsion may be formed by batching of ingredients in appropriate proportion. Molten oil blended to requirement is mesured into an agitated tank. Oil-soluble ingredients including lecithins, monoglyceride surfactants, oil soluble flavourants, oil soluble vitamins and colourant are added to the molten oil.
Pasteurized aqueous phases are typically prepared from milk, water, salt, water soluble flavourants and preservative. The aqueous phase is maintained at 5 -10ºC after pasteurization and before mixing with the oil phase. Addition of a cold aqueous phase to a warm oil phase is accomplished with continuous agitation to form a coarse, but very unstable emulsion. An alternative process is continuous blending of oil phase and aqueous phase using proportioning pumps or other metering devices.
The mixed oil and aqueous phases are pumped to a tube chiller or swept-surface heat exchanger. The tube chiller is generally a cylinder that is cooled externally and that contains a centre shaft to which are attached scraper blades. As the centre shaft is rotated, the scraper blades remove product from the interior anular surface of the heat exchanger. The liquid emulsion is pumped through the heat exchanger/chiller, resulting in super cooling of the fat by a temperature drop from about 40 - 46ºC to 7 -10ºC. Crystallization begins with cooling and continues for 24 hours or more. Crystal development increases the temperature of the margarine emulsion several degrees as heat of crystallization is released. Post-chilling working by a rotor-stator texturizer influences the texture of the product. Generally, tub margarines are mechanically worked to allow crystal growth while preventing formation of a firm crystal lattice. As working increases, product consistency softens.
Stick margarines, however, are generally allowed to rest briefly post-chilling and before packaging to allow firming of product to withstand the extrusion forces of stick making. Whipped margarines are produced by injection of nitrogen into the liquid emulsion for either tub or stick form.
The working and/or resting steps are used to obtain optimal packaging consistency as well as for optimization of performance characteristic. Packaging of tub margarine is generally accomplished by automatic filling into tubs. Stick margarine is produced by extrusion of chilled product into forming moulds with subsequent wrapping of moulded sticks. Margarines are generally refrigerated for maintenance of physical structure and flavour and to prevent spoilage by action of microorganisms.Margarine analogs have proliferated in recent years. Products similar to margarine but not conforming to requirements set forth in the appropriate legislation are normally termed spreads.
4. Spreads
Spreads are edible emulsion made in semblance of margarine, but that contain less fat. Typical fat content ranges from 40% to about 75%. These products are usually made with the same ingredients as margarine, although slightly more emulsifier is used. Lower-fat spreads are consumed primarily as table spread rather than as cooking agents. Spreads are marketed since 1975 and represent a growing segment of the yellow fat market. Manufacturing processes are as for margarines, but special surface active materials might be needed in order to permit the subsistance of a continuous fat phase even at relatively low total fat content.
5. Water-based emulsions
Water-based emulsions are important products both as spreads and as seasonings. The term mayonnaise has been used to describe these systems. Manufacture reposes on maintenance of the emulsion during shelf life and efficient surface active ingredients are needed for this purpose. Oil, being a discontinuous phase, does no play a dominant role in defining the rheological and structural properties, and its composition will be the result of nutritional factors as well as of availability and purpose. Emulsions are metastable states and the temperature domain within which emulsion subsists is relatively limited. Emulsions should therefore be stored away from strong heat and used at moderate temperatures.
6. Shortenings
Shortening is a term which designates a solid fat. The designation which is internationally accepted stems from the fact that fat can and indeed is used in bakery in order to diminish the total volume of finished product (i.e. in order to shorten the pastry). Shortenings are commonly used in industrial baking instead of margarines and they may be manufactured according to a wide series of different specifications, covering a wide variety of applications, as regards their SCI, color and additives.
4. Deterioration of oils, fats and emulsions
4.1. Physical, chemical, biochemical and biological degradation of fats and oils
Enzymatic Hydrolysis
Hydrolases
The ubiquitous presence of hydrolases in foodstuffs and their propensity to liberate short-chain acids (<C14) which have low odour and aroma thresholds, from the tasteless tryacylglycerol components means that this is an extremely important mechanism of introducing sensory alteration in food. Moreover, the fact that microorganisms also use hydrolases for food digestion means that also these will be actively involved in changing food properties. Changes due to hydrolysis may range from extremely desirable such as the soapy notes imparted upon blue cheeses by hydrolases originating from the mould, to extremely unpleasant high butyric notes in butter and lauric acidity in coconut oil.
In commonly used vegetable oils, the polyunsaturated C18 acids which are much better substrates than the tryacylglycerols from which they originate, will, when liberated by hydrolysis, readily be oxidised into very odoriferous compounds.
In fruits and vegetables and especially when tissues are sliced or homogenised during processing, oxidation and lypolysis will generally be very quick. Free acids are responsible for off flavours and enhanced oxidability. Also, enzymatic hydrolysis of a small amount of the acyl lipids present can not be avoided during disintegration of oil seeds, a characteristic which makes further processing mandatory for separation and/or denaturation of those enzymes.
Lipid hydrolysis is catalysed by some hydrolases, whose action and specificities differ. In particular, lipases will hydrolyse triacylglycerol and related moieties when emulsified and other hydrolases are specific for polar lipids.
Lipases
Lipases are active on a water/lipid interface and in this respect differ from esterase enzymes which cleave only water-soluble esters, such as triacetylglycerol.
Lipase activity is detected in, for example, milk, oilseeds (soybean, peanut), cereals (oats, wheat), in fruits and vegetables and in the diggestive tract of mammals. Many microorganisms release lipase-type enzymes into their culture media, and these may contribute to enhance the deterioration of foods.
As to specifity, fat-splitting enzymes, which preferentially cleave primary HO-group esters are distinguished from those which indiscriminately hydrolyse any ester bonds of acyl glycerols.
The 48 kcal lipase secreted by pig pancreas has probably been the best studied. It cleaves all acyl glycerols but preferred substrates are triacylglycerols, and worst of all monoacylglycerols, and in any case breaks the ester bonds at positions 1 or 3 only.
Oat and Aspergillus flavus lipases present no positional specificity whatsoever, whereas Geotrichum candidum lipase is specific for oleic and linoleic residues in any position, and Mucor miehei and Penicillium roqueforti lipases also show 1,3 specificity.
Acyl migration from position 2 to 1 is thermodinamically favoured and normally precedes enzymatic hydrolysis of that acyl residue; longer hydrolysis times are needed for completeness of this reaction unless an unspecific lipase is used.
When hydrolysis of emulsions is being studied, the size of oil droplets in as much as it affects the total oil/water interfacial area, the larger the oil/water interface and, therefore, the higher the apparent lipase activity. Interfacial area should be considered when substrate emulsions are used in the assay of enzyme activities.
Activity of lipases at the interface is explained by assuming that part of the lipase molecule is hydrophilic and part is hydrophobic, hence the enzyme sits ot the interface and the active site lies next to it. Hydrolysis of the ester bond occurs with the involvement of Serine, Histidine and Aspartic residues as in chymotrypsin peptide bond breaking. Lipase has a leucine residue within the active site in order to maintain a hydrophobic interaction with the hydrophobic lipid and bring it to the reaction centre, whereas serine proteinases do not have such a residue because their substrates differ.
Lipases with high specificity may be used to taylor food ingredients. An example may be found in the production of CBS, which are replacements for cocoa butter, and in the production of high polyunsaturate oils which may have nutritional advantages. Another example is the use of unspecific lipases when randomization of acyl groups is to be obtained as for instance in enzyme mediated interesterification for the production of margarines and spreads.
Phospholipases
Some of the enzymes that cleave phosphatides are specific, and indeed have been invaluable tools for the elucidation of the three dimensional structure of lipids. Their preferred mode of action is shown schematically below:
Phospholipases A1 and A2: Both enzymes occur in many mammals, and both yield lysolecithins. They differ in the acyl group which they will cleave off.
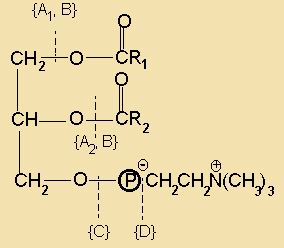
Phospholipase B: is specific for removing the acyl residue which is left in a lysophosphatide, and also ocurrs in mammals.
Phospholipase C: will hydrolyse lecithin to a 1,2-diacylglycerol. It does not ocurr in mammals, only in bacteria , and also in some snake venoms.
Phospholipase D: which is widespread in cereals and oilseds,cleaves the choline group in the presence of water or an alcohol, yielding phosphatidic acid, free or esterified.
Glycolipid hydrolases
Enzymes able to cleave the acyl residues from mono- and digalactosyl-diglycerides are obtainable from green plants. Such an enzyme obtained from potato also acts against other polar lipids and successfully removes the acyl residue from monoacylglycerols and lysolecithins, but is inactive against triacylglycerol moieties.
Oxidation of Unsaturated Acyl Lipids
The oxidative deterioration of food items proceeds either through the peroxidation of unsaturated components in fats and oils or through the catalytic conversion of phenolic groups into quinones and their coupling and polymerisation.
The latter reactions are obvious and may best be observed in cut apples, they are mediated by phenolase enzymes and generally called enzymatic browning reactions. They are delayed by low pH, ocurr at relatively high water activity and will not be studied here. The former, who will be addressed below, may happen under a variety of circumstances, and may generally be said to be favoured by light, transition metal ions, high temperature and low water activity.
As oxidations will exhibit some dependency on oxygen availability. Some of these reactions may be catalysed by enzymes, but many are not. They strongly affect the aroma and odour of foods, both agreeable and disagreable (rancid) notes being produced, depending on the circumstances. The low odour thresholds of the intermediate polarity products which are the result of these reactions render them detectable shortly after their onset.
One may distinguish various types of lipid oxidation, depending on the nature of the agent(s) responsible for its onset and rate. Thus autoxidation and lipoxygenase catalysis are both effective in yielding hydroperoxides.
Induced changes in food aroma are continually assesssed by consumers as objectionable, for example, as rancid, fishy, metallic or cardboard-like, or as an undefined aged, stale or warmed up flavour. On the other hand, the fact that some volatile compounds, at a level below their off-flavour threshold values, contribute to the pleasant aroma of many fruits and vegetables and to rounding-off the aroma of many fat- or oil-containing food should not be neglected.
Autoxidation
The term autoxidation designates a complex set of reactions which result in the fixation of oxygen by lipids and shows complex autocatalytic behaviour and involves a number of interrelated reactions of intermediates. The observation that saturated lipids only autoxidise very slowly means that this reaction occurs involving the isolenic unsaturated double bonds present. Autoxidation of food is usually modelled by that of a simpler system in which either a triacylglycerol with three equal acyl groups or a simple ester are used as models in the presence of oxygen under controlled experimental conditions, and the products obtained at various stages and rates of interconversions observed are studied.
These studies have shown that many variables can affect autoxidation rate. Thus fatty acid composition, light, transition metal ions, oxygen pressure, presence of antioxidants, prooxidants, temperature, moisture content and distribuition were shown to affect the rate of the reaction.
It may be stated that this reaction, though still not completely defined, seems to occur at an increasing rate, after an initial stage called an induction period.
Autoxidation leads to products with higher oxygen content. Some of these are more volatile and their low sensory threshold conveys an early warning of their presence. Others are polar compounds such as acids and they will confer to the oxidising food a higher conductivity. Still another range of products are polymeric.
The polar compounds form the basis of detection in a method for measuring oxidative stability of oils. In this method, which is only one of a huge series of methods - confirming the idea that autoxidation is complicated - a sample of oil is subject to high temperature (say 100ºC) in a vessel which operates under moderately high oxygen pressure, and conductivity is measured by way of electrodes. The induction period before quick rise in conductivity measures the oxidative stability of the oil in this so called "Rancimat" method.
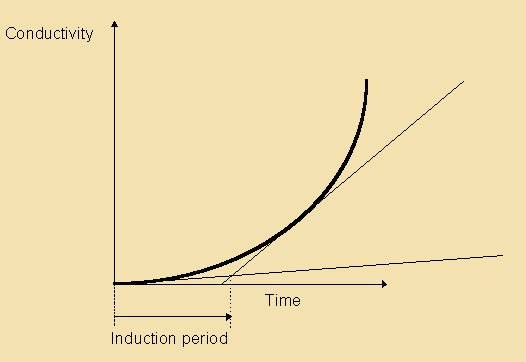
Induction period and realtive rate of oxidation for fatty acids at moderate temperatures is strongly dependent on unsaturation. An approximate idea of relative reactivities and induction periods (which must be considered as a mean value because factors such as light intensity, photosensitizers and antioxidants would alter these relative values for oils), is given below:

Both the induction period and the rise in the reaction rate in the series oleic, linoleic and linolenic acid can be explained assuming that oxidation proceeds by a sequential free radical chain-reaction mechanism. Relatively stable radicals that can abstract H-atoms from the allylic methylene groups in olefinic compounds are formed. The oxidation process is therefore a radical-induced chain reaction which may be divided into the classical steps of initiation, propagation, branching (and this one explains the autocatalysis) and termination. Initiation results in the formation of free radicals P< by a variety of processes, and this is able to generate an alkyl radical E<.

The reaction rate constants for the different steps of this radical chain reaction markedly differ in magnitude, especialy because of the stability of the peroxy free radicals (EOO·.) which reacts so slowly that it conditions and limits the overall oxidation rate. Thus formation of monohydroperoxide molecules (EOOH) which is achieved by abstraction of an H-atom from a fatty acid molecule, is the slow process in the formation of radicals.
Branching, shown in reactions 6 and 7 above, is responsible for the autocatalytic effect, in as much as it increases the number of reacting chains, and conversely, termination diminishes the number of reacting chains. it is obvious that these later reactions only happen when the number or radical species is high, for they depend upon the fact that two of these meet before either finds another adequate substrate.
Branching as shown in reaction 7, though exothermal in contrast to reaction 6, only becomes significant when hydroperoxide concentration is likewise high, a condition which normaly is fulfilled only long after the food item is no longer considered edible. Reaction 6 will be catalysed by transition metals and their complexes, which in this way act as prooxidants since the radicals produced in these reactions can start chains anew.
Each new radical can start a chain responsible for many molecules of hydroperoxide, before a termination reaction stops it, and if there is air enough it will keep adding to the allylic positions. In a high oxygen atmosphere, such as air, termination by reaction [9] of two peroxy radicals is more probable. The resulting species has a labile tetraoxygen group which decomposes rapidly yielding singlet oxygen and two new chains, and is therefore not a pro bone termination recation at all, for singlet oxygen has oxidising capabilities which far exceed those of triplet oxygen.
![]()
Termination reactions depicted as [8] and [10] above play a role when the oxygen level is low, as may happen in the inner portion of a fatty food, or under high temperature conditions..
The whole scheme presented so far is an accurate description of phenomena at the early stages of autoxidation. Nevertheless more must be said about initiation, and also about the fate of peroxides formed and the importance of the role of their decomposition or further oxidation products with the initial products of autoxidation.
Monohydroperoxides
Of the various radicals involved in autoxidation, the peroxy radical is much more stable than the other alkoxy, hydroxy or alkyl radicals. It is thus quite able to select a hydrogen atom from an allylic group (DR-H=322 kJ/mole) or even from a bi-allylic position as in linoleic or linolenic moieties (DR-H=272 kJ/mole) instead of the much more inaccessible alkyl radical which show dissociation energies above the 400 kJ/mole mark.
That these energy differences matter is shown by the difference in reaction rates and induction periods observed at moderate temperatures when linolenic (two bi-allylic positions) or linoleic (one bi-allylic position) are suffering autoxidation, relative to that shown by oleic (one allylic position) or even to the stability of stearic moieties.
The peroxy radical formed during propagation is slow reacting and therefore it selectively abstracts the most weakly bound H-atom from a fat molecule. It differs in this property from, for example, the substantiallly more reactive hydroxy (HO· ) and alkoxy (RO· ) radicals.
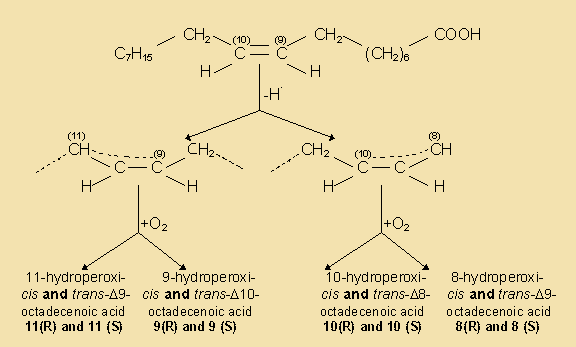
As may be seen from the schematic depictions, an oleic substrate will actually yield as result of the first autoxidation stage a mixture of 8-, 9-, 10-, and 11- hydroperoxides. If both geometrical (cis-trans) and stereochemical (R-S) isomerism is taken into acount - as of course it should be - this means that sixteen different hydroperoxides are produced. Inspection of the diagram shows that it is after abstraction of one hydrogen atom that the bond between positions 9 and 10 loses its character as a double bond, hence becoming prone to rotational isomerisation to a cis geometry, thermodinamically more stable. When the double bond migrates, the general case is that the trans isomer be predominant, say twice as abundant as the cis .
The methylene group in position 11 is the initial site for the abstraction of an H-atom in oxidation of linoleic acid, because it is the only bi-allylic position. The pentadienyl radical generated will after reaction with oxygen yield a conjugated hydroperoxidiene system with oxygen at positions 8 or 13 and migration of the double bond nearest to it, rather than that which would result from substitution at the original position 11 or those in which this position remains an unchanged methylene group. Geometric isomerism might or might not occur with double bond migration, and a number of isomers will therefore result.
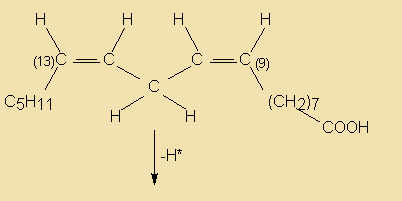
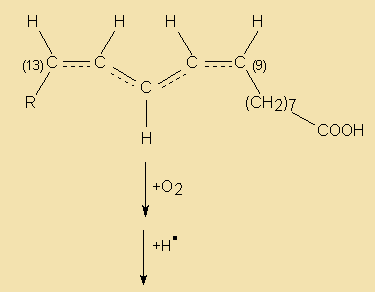
The hydroperoxides have a UV maximum absorption at 235 nm and can be resolved by high performance liquid chromatography of their methyl esters, either directly or after reduction to hydroxydienes.
Nevertheless, the monoallylic groups in linoleic acid (positions 8 and 14 in the molecule), in addition to the bi-allylic group (position 11), also react as allylic systems to a small extent, yielding hydroperoxides (8-, 10-, 12- and 14-OOH), each of them a mixture of cis and trans isomers with two isolated double bonds. These minor monohydroperoxides only amount to ca. 4% in total.
The hydroperoxides resulting from autoxidation (3O2) and photoxidation (1O2) of unsaturated fatty acids are shown in table form:
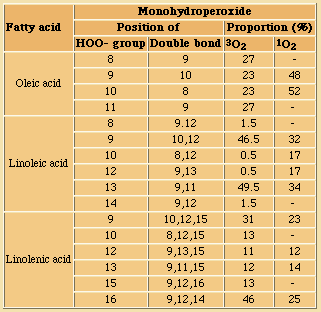
In an entirely similar way, it can be seen that autoxidation of linolenic acid yields four monohydroperoxides (stereochemical and geometric isomerism not included). Formation of the monohydroperoxides is easily achieved by H-abstraction from the bi-allylic positions 11 and 14. Each of the resulting pentadiene radicals will then suffer addition of oxygen and of a hydrogen atom as shown for linoleic acid. However, the positional isomers are not formed in equimolar amounts; a clear predominance of the 9- and 16- isomers is apparent in the table above. The configuration of the conjugated double bonds again depends on reaction conditions. Cis-hydroperoxides are the main products if the reaction is performed at temperatures below 40ºC.
Reactions involving b-fragmentation and cyclisation may compete with hydrogen abstraction by peroxy radical, thus keeping the conversion to hydroperoxide from being fully effective. Moreover, both allyl peroxy radicals and hydroperoxides can undergo 1-3 rearrangement via a b-fragmentation mechanism, and after a new bond to oxygen is made, in a positional isomer of the starting material, in a process which is akin to internal return.
Further oxidation
Peroxy radicals with ![]() ,
, ![]() double bonds which mainly originate in singlet oxygen attack to polyunsaturated fatty acids but also from the above mentioned rearrangement of autoxidation adducts, may undergo cyclisation reactions in competition with reactions leading to monohydroperoxides. The resulting epidioxides may suffer oxygen addition and abstraction of a hydrogen atom yielding hydroperoxide-epidioxides .
double bonds which mainly originate in singlet oxygen attack to polyunsaturated fatty acids but also from the above mentioned rearrangement of autoxidation adducts, may undergo cyclisation reactions in competition with reactions leading to monohydroperoxides. The resulting epidioxides may suffer oxygen addition and abstraction of a hydrogen atom yielding hydroperoxide-epidioxides .
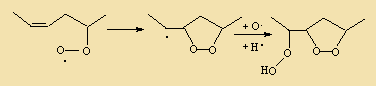
Peroxy radicals with isolated ![]() ,
, ![]() double bonds are formed as intermediary products after autoxidation and photooxidation (reaction with singlet O2) of unsaturated fatty acids having two or more double bonds. Fragmentation occurs when a hydroperoxide-epidioxide is heated, resulting in the formation of aldehydes and aldehyde-acids.
double bonds are formed as intermediary products after autoxidation and photooxidation (reaction with singlet O2) of unsaturated fatty acids having two or more double bonds. Fragmentation occurs when a hydroperoxide-epidioxide is heated, resulting in the formation of aldehydes and aldehyde-acids.
Peroxy radicals interact rapidly with antioxidants which may be present to give monohydroperoxides. Thus, it is not only the chain reaction which is inhibited by antioxidants, but also ![]() -fragmentation and peroxy radical cyclisation.
-fragmentation and peroxy radical cyclisation.
Initiation of radical chain reactions
The knowledge of the reactions which predominate during the induction period is important because it may help find out how to make it last longer, and thus improve the resistance of oils to oxidative degradation.
Two different types of reactions have been recognized as important in this process.
The first of these has to do with the initiation reactions which are responsible for by-passing the energy barrier required for oxidation of allylic groups and includes photosensitised oxidation (photooxidation) and lipid oxidation by lipoxygenase catalysis.
Both of these provide initial hydroperoxides, which are further converted into radicals by reactions of a second type.
Heavy metal ions and heme groups may be involved in reactions of this second type.
Enzyme catalysed reactions generating the superoxide radical anion can be placed in between theses two types of reactions, for they need also H2O2 to be able to initiate radical reactions.
A.Photooxidation
In order to understand photooxidation, or light promoted oxidation, and to differentiate it from autoxidation, it should be understood that the oxygen ground state is a triplet, but that an excited singlet is available with an energy only 92 kJ/mole above that of the ground state.
Whereas triplet ground state oxygen tends to react as a diradical, using its semioccupied orbitals for the purpose of building new bonds and preferring other radicals as substrates, the excited singlet may use wholy occupied and/or empty orbitals for the same purpose, hence behaving as an nucleophile/electrophile and participating in electrocyclic reactions and reacting with other molecular entities.Hence, in the reaction with oleic acid, the singlet state oxygen attacks the 9-10 double bond producing an equimolar mixture of the 9- and 10- hydroperoxides (R and S).
Light can trigger lipid oxidation in two different ways, both mediated by small amounts of compounds called sensitisers.
The first type of photosensitiser, Type I sensitisers (S), once activated by light (S*), reacts directly with a substrate, generating radicals which are the initiators of the oxidation process.
The other sensitisers are those which activate the ground state of oxygen to the first singlet state, and they are called Type II photosensitisers.
Photooxidation processes involving both types of sensitisers occur simultaneously, and both the structure and availability of the sensitisers present as well as the concentration and structure of the substrate available for oxidation are involved in determinig which one will prevail.
As the hydroperoxide isomer distribution resulting from oxidation with singlet and triplet oxygen differ, it is possible to determine the relative importance of these two mechanisms by product analysis.
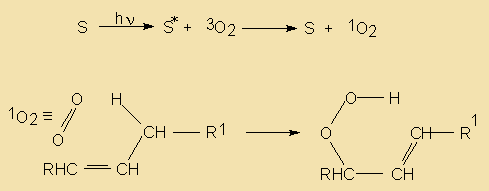
Thus, one can distinguish Type I from Type II photooxidation, and in this way, prove that sensitisers such as chlorophylls, pheophytins, and riboflavin, commonly present in food items will catalyse the Type II oxidation of compounds containing unsaturated acyls.
The formation of hydroperoxides exclusively at former double bond (sp2) carbon atoms of unsaturated fatty acids may be seen in the table above, the actual formulas of the four (S,R) racemates formed evidencing also the preferential cis-trans isomerisation which occurs when linoleic acid is oxidised by singlet oxygen. It may be seen that, in addition to the two hydroperoxides with conjugated diene system, two others are obtained with isolated double bonds.
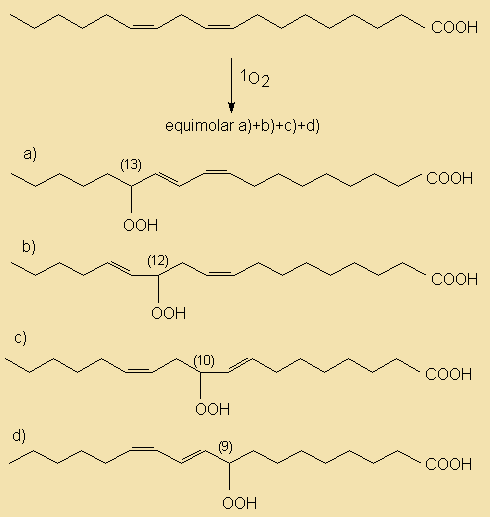
Reaction of singlet oxygen (1O2) with double bonds is inhibited by carotenoids present, for these compete for the 1O2 depriving it of its excess energy and making it revert to the ground state triplet, whereas the carotenoid itself becomes a triplet but dissipates excess energy thermally and reverts to a singlet. This quenching effect is very fast (k = 3 x 1010 mole-1s-1), therefore carotenoids are particularly suitable for protecting fat (oil)-containing food from Type II photooxidation. Their role as auxiliary pigments in photosynthesis means they ubiquitously accompany chlorophyll, whose prooxidant effect they effectively tend to nullify.
B.Heavy metal ions
Fats, oils and foods always contain traces of heavy metals, the complete removal of which in a refining step is not industrially performable. These ions, primarily Fe, Cu and Co, may originate from impurities accompanying the plant material, packaging of oilseeds or oil, or from processing equipment. These ions are responsible for initiation reactions of the second type, by catalysing the decomposition of hydroperoxides into radicals which initiate new radical chains in the oxidation process.
Vegetable oils of the linoleic acid type, such as sunflower and corn germ oil, which are easily oxidisable, should contain less than 0.03 ppm Fe or 0.01 ppm Cu to ensure acceptable stability. This may be as high as 5 ppm for both Cu and Fe in animal fats with a high content of oleic and/or stearic acid.
The presence of a hydroperoxide group is a prerequisite for metal ion activity as an oxidation catalyst leading to hydroperoxide decomposition and new radical chain initiation:
![]()
The first of these reactions is much faster than the second, hence the alkoxide radicals more important initiators than the peroxide. Water phase antioxidants such as ascorbic acid can prove to have here a prooxidant effect for they tend to reduce oxidised metal ions to their lower oxidations states, hence permitting more of the first reaction above to take place. In this case there is also a pH effect, but normal values for food are near-optimal for metal ion redox catalysis of peroxide decomposition.
In any case, it is important to exclude hydroperoxides or diminish their concentration.
Last but not least water activity will condition autoxidation. Both very low and very high water activity values seem to promote food oxidation, the minima occuring at intermediate aw values of 0.25-0.3.
At high water activity prooxidant mobility (including enzymes) is the probable reason for this, and at very low water activity, increased oxygen mobility permited through positions left vacant by removed water molecules.
C.Heme compounds
Proteins exhibiting heme-like prosthetic groups are ubiquitous and an indispensable part of the electron transport chain. They can chelate peroxides cleaving them into an alkoxy and a hidroxy radical and then releasing them, especially after having been denatured in order to expose the heme groups. this effect is not dependent on pH or added ascorbic acid as it does not involve an alteration of the oxidation state of the metal ion.
Denaturation of lipoxygenase which is the main enzymatic culprit for oxidation of vegetable foods must be done with care not to denature catalase or peroxidase too strongly, as this may expose their heme groups which will become oxidation catalysts.
D.Activated oxygen from enzymatic reactions
As part of the biochemical mechanisms entailing the use of oxygen, an enzyme cascade is involved. These reactions entail the formation of a series of progressively more reduced species, in an aqueouis environment. Thus if oxygen, or rather dioxygen is the most oxidised form, and water is the reduced form, these intermediate forms are, in first place, the superoxide anion, which is the result of a one electron reduction of dioxygen, and behaves as a nucleofile, and its protonated form the hydroperoxide radical which only is formed under conditions rather more acidic than physiological ones.
Superoxide generated by the flavin enzymes will slowly dismutate into dioxygen and hydrogen peroxide, a reaction which can also be catalysed by superoxide dismutase. Hydrogen peroxide can suffer photochemical or chemical decomposition yielding the extremely hard and reactive hydroxyl radical, unless it is first reduced to water by catalase. A particularly efficient way of generating the hydroxyl radical is the so-called Fenton reaction, whereby a transition metal ion complex catalyses the oxidation of superoxide to dioxygen while generating the hydroxyl radical from hydrogen peroxide:
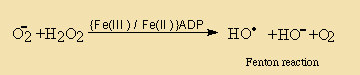
The hydroxyl radical formed will initiate autoxidation.
Reactions of primary oxidation products
The hydroperoxides formed in the primary double bond oxidation process of unsaturated acids and lipids are not stable, but they also do not possess any noticeable sensory properties. They will nevertheless promptly react under normal autoxidation conditions yielding a series of compounds which have extremely low sensory detection thresholds. These mainly comprise carbonyl compounds and a few hydrocarbons, but substituted furanes and alcohols are also formed.
Main volatile carbonyl compounds from unsaturated fatty acids after uptake of 12 mole oxygen, (in ppm).
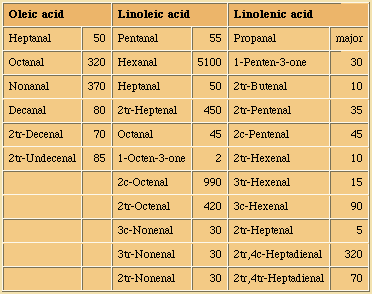
The odour threshold values of these compounds are low or even very low, and can be lower in a polar medium than in a lipid phase where their solubility is high.
The autoxidation of a-linolenic acid produces especially strong odours, and it is therefore extremely easy to spot early.
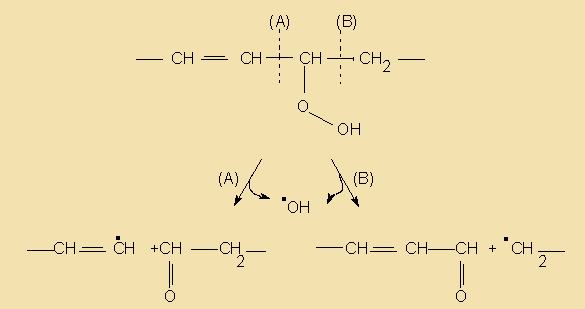
Of these one should stress 3-cis-hexenal and 2-trans,6-cis-nonadienal. Other carbonyl compounds with similar aromatic strengh and low threshold will be released from animal fats contributing to a characteristic "warmed-up" flavour, or when processing and especially if re-processing vegetable oils.
The ![]() -scission of hydroperoxides catalysed by heavy metal ions or heme compounds is the main pathway explaining the formation of these carbonyl compounds. Of the two different mechanistic possibilities (A) and (B), shown in the figure above, the second one is energetically preferred, the transition state leading to it presumably lower in energy than that involved in (A), as might have been hinted at by the fact that a conjugated carbonyl compound is obtained through (B). Though some of the structures of products obtained may be rationalised in this way, as is the case for 2,4-decadienal and pentane can be explained in this way, this is not the case for hexanal, whose preferential formation in aqueous phases may point to an ionic mechanism.
-scission of hydroperoxides catalysed by heavy metal ions or heme compounds is the main pathway explaining the formation of these carbonyl compounds. Of the two different mechanistic possibilities (A) and (B), shown in the figure above, the second one is energetically preferred, the transition state leading to it presumably lower in energy than that involved in (A), as might have been hinted at by the fact that a conjugated carbonyl compound is obtained through (B). Though some of the structures of products obtained may be rationalised in this way, as is the case for 2,4-decadienal and pentane can be explained in this way, this is not the case for hexanal, whose preferential formation in aqueous phases may point to an ionic mechanism.
The further oxidation reactions of monohydroperoxides and carbonyl compounds are among the possibilities of explaining some of the products obtained and which cannot be obtained through (B).
Sensory properties of aliphatic aldehydes and vinyl ketones
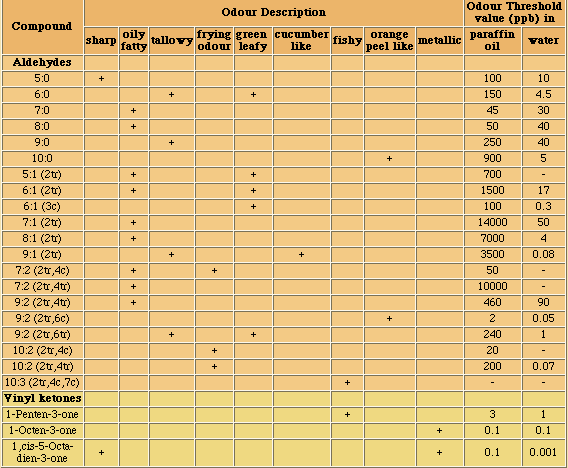
That tandem oxidation processes are ocurring is supported by the fact that 2-alkenals and 2,4-alkadienals suffer oxidation at faster rate than the parent unsaturated fatty acids. The autoxidation of 2,4-decadienal yields hexanal and other volatiles which fit into the pattern obtained from linoleic acid oxidation. Saturated aldehydes will be more stable and eventualy predominate in the mixture.
Other compounds such as pentanal need as precursors the oxygen heterocycles which may result from peroxide intramolecular rearrangements.
The autoxidation of fatty acids with three or more double bonds invariably leads to high yields of malondialdehyde. This compound may serve as a protein denaturing reagent by crosslinking with two amino groups. Malondialdehyde may result from alinoleic acid, and its presence is used as an indicator for fat or oil oxidation (TBA -thiobarbituric acid test) where a colour producing reaction is used as an indicator.
Several furan derivatives occur among the autoxidation products of linoleic and linolenic acids. These derivatives may be involved in the development of an off flavour specific to soyabean oil, removable in the refinery but which may come back if oxidation is allowed to start again, in a phenomenon termed flavour reversion.
The main hydrocarbon constituents of the volatile hydrocarbon fraction, ethane and pentane can easily and quantitatively be detected by headspace chromatography and are used as indicators of lipid autoxidation.
Lipoxygenase
Lipoxygenases (linoleic acid oxygen oxidoreductase) are ubiquitous. Present in many vegetable and animal cells. Lipoxygenases catalyse the oxidation of 1-cis-4cis unsaturated fatty acids to the hydroperoxides, as happens during autoxidation, but will not oxidise with oleic acid.
Lipoxygenases are metaloproteins with an Fe atom in its active center. The enzymes are activated by hydroperoxide and the reaction pathway always includes the migration of one double bond in order to yield a conjugated system, which is normaly a hydroperoxide but may also be a peroxy radical. These reactions occur at substrate and pH-dependent rate even at ror below room temperature and in the dark.
Enzyme-catalysed oxidation is initiated even in the absence of hydroperoxides. This means the enzyme alone is able to overcome the energy barrier of this reaction, and therefore it has to be thermally inactivated if the reaction is not to be allowed to proceed.
At least two fundamentally different types of lipoxygenase enzymes exist in plants. Type I lipoxygenase oxidises only free fatty acids and will do so with a high stereo- and regioselectivity giving rise to an optically-active hydroperoxide with a conjugated cis-trans-diene system, with the function at either end of the pentadiene system. It will, for instance, form preferentially either 9- or 13-hydroperoxides (R, or S) from free linoleic acid as the substrate.
Occurrence and properties of various lipoxygenases
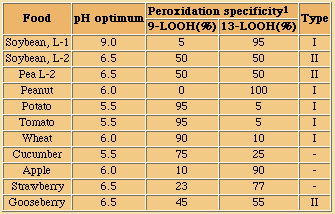
Type II lipoxygenase, on the other hand, will yield both 9- and 13-hydroperoxides (as in the noncatalysed autoxidation), and it also reacts with esterified substrates, thus attacking oils and fats without requiring prior release of fatty acids by a lipase enzyme.
This enzyme will also oxidise carotenoids and chlorophyll and thus degrade them to colourless products, a property used in flour "bleaching". This can be performed by the addition of , for instance, a small quantity of uncooked mown soyabean to the dough. This activity may be rationalised if the possibility that this enzyme may release alkylperoxyl radicals, and not only hydroperoxides, into solution, and these attack the pigments.
The type II enzyme present in legumes will when in contact with lipid substrates, yield a mixture of aldehydes identical to that obtained during autoxidation.
High temperature degradation of oils and fats:
When subject to high temperature as during cooking and especially frying operations, reactions of oils with food components can greatly accelerate oxidation. The reactions of oils and fats with other food components under these circumstances will be addressed with the degradation of emulsions.
Deterioration of heated oils, as may be seen in oil used for multiple frying operations, is characterised by development of a dark colour and settling of a polymeric dark mass. During frying operations, high oil temperature and water vapour conjure to make oxygen pressure very low. Deterioration will ensue thanks to partial oxidation, but most low molecular weight compounds formed will be steam volatilised and only those with low vapour pressure will accumulate. These include fatty acids and high molecular weight polymeric dark brown material.
Reactions involved in these transformations include fragmentation, mainly by b-scission after hydroperoxide formation, leading to volatiles responsible both for the initially pleasant notes in frying and later for the unpleasant odour of spent frying oil, as described, and also to cyclization reactions, both intra- and intermolecular, occuring via electrocyclic transition states, such as the Diels-Alder and -Ene reactions.
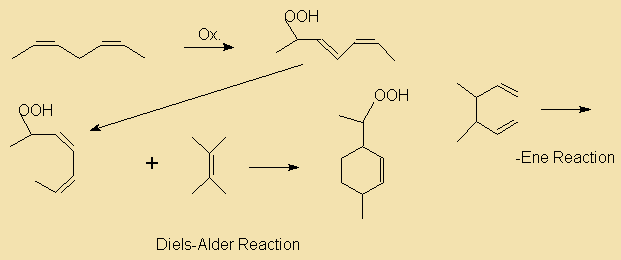
These reactions and other similar ones may explain the appearance of decomposition products with cyclic structures. Polymers with ether or peroxide bonds may also appear due to reaction of alkoxide or alkylperoxide radicals directly with alkyl or alkoxide radicals, especially in the later stages of oxidation.
The ![]() -scission reaction may leave carbonyl groups attached to the triacylglyceride moiety (core aldehydes). Core aldehydes are non-volatile and extremely reactive species, capable of adding to amine groups from protein material (available from other components of food) and starting processes which eventually lead to policyclic compounds, which may be heterocyclic and will contribute to the light absorption pattern of the polymeric mass. Total solids content is one of the criteria for discarding a cooking oil, especially because this may be determined experimentally with some ease. It has therefore been used in many countries as a legal criterium limiting appropriateness of an oil for frying.
-scission reaction may leave carbonyl groups attached to the triacylglyceride moiety (core aldehydes). Core aldehydes are non-volatile and extremely reactive species, capable of adding to amine groups from protein material (available from other components of food) and starting processes which eventually lead to policyclic compounds, which may be heterocyclic and will contribute to the light absorption pattern of the polymeric mass. Total solids content is one of the criteria for discarding a cooking oil, especially because this may be determined experimentally with some ease. It has therefore been used in many countries as a legal criterium limiting appropriateness of an oil for frying.
Eventually the foaming from the frying oil increases and smoke is liberated upon heating even at low temperatures. At this stage the use of oil in frying is no longer only a long term health risk but also represents, especially if using an open flame, a dangerous behaviour because of it's increased flamability and the risk of fire.
4.2. Physical, chemical, biochemical and biological degradation of emulsions
Degradation of emulsions presents similarities and differences to that of oils. Emulsions are metastable states, and therefore will suffer irreversible damage when heated or cooled below the temperature domain within which they maintain their metastability. This is an instance of degradation attributable to a physical cause.
Chemical and biochemical degradataion of emulsions has been dealt with in various sections already.
Chemical degradation by hydrolysis at the interface is not very important, because this uncatalysed reaction is extremely slow. On the other hand, surface active agents will in principle be occupying this interface, and might even provide physical barriers to species which would react with components of the inner phase. Chemical oxidation of emulsions can be a very taxing phenomenon indeed, and those whose inner phase is hydrophobic (i.e. o/w) are especially prone to it. The water phase acts as a good carrier of oxidising species to the interface, and therefore it is immediately at this interface that oxidation will occur. The surface active agents present at the interface may help delay oxidation by lessening the rythm of mass transfer of oxidants, but may provide reactive centers at a later stage of this phenomenon. Thus, carbonyl compounds formed from peroxidised fatty acids will easily react with amino compounds from protein present at the globular interface as a surface active ingredient. The resulting chemically altered protein may loose some of its surface active properties and its isoelectric pH will change. As more and more of its amino groups are used in such a fashion, the protein will complete chemical denaturation.
Biochemical degradation by the action of lipase enzymes is especially important and occurs at the interface as mentioned. Other surface phenomena which will occur include oxidation mediated by oxidising enzymes also mentioned earlier.
Protection against oxidation may be provided by antioxidants as well as by a series of good manufacturing practices. These will be dealt with below, but an important phenomenon, termed "the polar paradox", will be mentioned straight away. This refers to the fact that against what would intuitively be thought, the most effective antioxidants in dispersions are those whose solubility is higher in the dispersed phase, hence tocopherols over ascorbate in o/w emulsions and ascorbate over tocopherols in the w/o systems.
4.3. Protection of lipids against deterioration
Protecting lipids against autoxidation is important albeit a little oxidation is necessary for the appearance of the pleasant notes in deep-fryed products.
Protection of lipids against oxidation may always be obtained by storage in the absence of oxygen, and also by avoiding undues exposure to high temperatures or light, preferably after blanching if lipoxigenase was present.
Natural protection of lipids may be afforded by antioxidants present in foods, or added to that effect.
These antioxidants may be of three different kinds, in respect to the molecular mechanisms of their protective activity. Artificial antioxidants may also be used in foods in order to enhance their oxidative stability.
4.4. Synthetic Phenolic Antioxidants
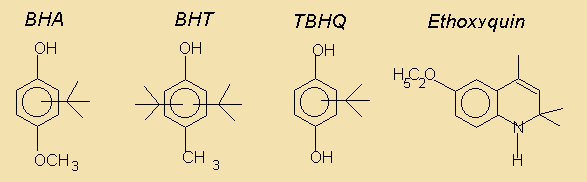
A first class of antioxidant species are those which effectively contribute to lower transition metal activity. These act generally by chelation and the better known are citric acid, phosphoric acid and some of its derivatives. These substances act not as antioxidants per se but rather as synergiistic ingredients which effectively diminish the rythm of chain initiation.
Antioxidants which act by interrupting a radical chain once started may be water soluble, such as ascorbate (Vitamin C) and its stereoisomer erithorbate, or oil soluble, such as the tocols (tocopherols and tocotrienols, of which a-tocopherol is Vitamin E)), the flavanones and flavonols present in vegetables and herbs and very abundant in tea, and the synthetic antioxidants BHT, BHA, TBHQ, or ethoxyquin, and other polyphenols such as gallic acid derivatives which occur in nature.
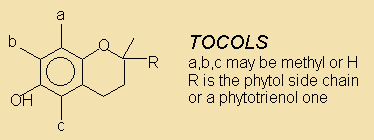
The latter category are also known as phenolic antioxidants, and their antioxidative capability seems to bear a relationchip not only to the number of phenol groups occupying 1,2 or 1,4 positions in an aromatic ring, but also to the volume and electronic characteristics of the ring substituents present Antioxidant activity may also be shown by quinonoidal systems as that present in phytomenadione (Vitamin K).
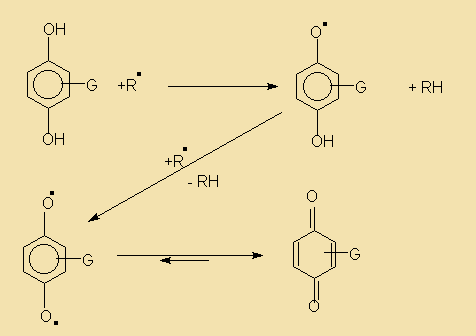
Antioxidant effectivity may be measured as the ratio of the induction period under a certain set of experimental circumstances (say 100 ºC, p(O2)=1 atm), for a given concentration, normally expressed as ppm (w/w), in a specified oil or fat.
If the protection factor for an antioxidant is Af , it may therefore be calculated as the ratio of induction period If when antioxidant is present and Io when it is absent.
Af=If/Io
Varying antioxidant concentration or mixing different antioxidants in different proportions may be performed to obtain data on synergies, antagonisms and optima.
References
The following four texts are general references for anyone wishing to dwelve into oils and fats. The first and third of these were instrumental in sections 3 and 4, and the last one in sections 1 and 2, whereas the second one was mostly inspirational.
• Belitz, H. - D.; Grosch, W.; "FOOD CHEMISTRY", Spinger Verlag, 1987.
• Coultate, T. P.; "FOOD, The Chemistry of Its Components", Royal Society of Chemistry Paperbacks, Second Edition, 1989.
• Fennema, Owen R.; "FOOD CHEMISTRY", Marcel Dekker, Inc., Second Edition, 1985.
• Hui, Y. H.; "BAILEY’S INDUSTRIAL OIL & FAT PRODUCTS", John Wiley & Sons, Inc., 1996.
The following are two selected recent references for readers wishing to pursue some matters in greater detail:
• W. Hamm, "Trends in edible oil fractionation", Trends in Food Science and Technology, April 1995, 6,121-6.
• E.F.Frankel, "Natural and biological antioxidants in foods and biological systems. Their mechanism of action, applications and implications", Lipid Technology, July 1995, 77-80.
Biofuels
Biofuels Library
Biofuels supplies and suppliers
Biodiesel
Make your own biodiesel
Mike Pelly's recipe
Two-stage biodiesel process
FOOLPROOF biodiesel process
Biodiesel processors
Biodiesel in Hong Kong
Nitrogen Oxide emissions
Glycerine
Biodiesel resources on the Web
Do diesels have a future?
Vegetable oil yields and characteristics
Washing
Biodiesel and your vehicle
Food or fuel?
Straight vegetable oil as diesel fuel
Ethanol
Ethanol resources on the Web
Is ethanol energy-efficient?
